
Abstract
In the past decade, the high throughput and low cost of sequencing/genotyping approaches have led to the accumulation of a large amount of data from genome-wide association studies (GWASs). The first aim of this review is to highlight how post-GWAS analysis can be used make sense of the obtained associations. Novel directions for integrating GWAS results with other resources, such as somatic mutation, metabolite-transcript, and transcriptomic data, are also discussed; these approaches can help us move beyond each individual data point and provide valuable information about complex trait genetics. In addition, cross-phenotype association tests, when the loci detected by GWASs have significant associations with multiple traits, are reviewed to provide biologically informative results for use in real-time applications. This review also discusses the challenges of identifying interactions between genetic mutations (epistasis) and mutations of loci affecting more than one trait (pleiotropy) as underlying causes of cross-phenotype associations; these challenges can be overcome using post-GWAS analysis. Genetic similarities between phenotypes that can be revealed using post-GWAS analysis are also discussed. In summary, different methodologies of post-GWAS analysis are now available, enhancing the value of information obtained from GWAS results, and facilitating application in both humans and nonhuman species. However, precise methods still need to be developed to overcome challenges in the field and uncover the genetic underpinnings of complex traits.
Similar content being viewed by others


Introduction
One of the powerful and widely used methods to detect associations between phenotypes and genetic variants is the genome-wide association study (GWAS), which analyzes genetic variants in common diseases. This method has been proved useful through an extreme increase in published GWAS results over time from its introduction (~2008) to the present (MacArthur et al. 2017). Some studies have recently reviewed novel techniques and methodologies for data pre-processing and GWAS methodologies, which increase the power of the analysis and help achieve accurate results from GWAS (Mortezaei and Tavallaei 2021; Tam et al. 2019).
Although GWASs can identify disease mechanisms, leveraging the wealth of GWAS-implicated loci and inferring truly causal variants is the main bottleneck that leads to gaps between genetic studies and therapeutic applications (Schaid et al. 2018). To close these gaps, post-GWAS pipelines have been developed (Box 1). Based on cell culture-based experiments and biological post-GWAS functional studies, candidate causal variants can be identified, and genetic variants in haplotypes associated with diseases can be defined. Post-GWAS analysis can identify genes functionally related to specific diseases and more quickly connect the functional part of the genome with clinical applications (Lin et al. 2018); for instance, the post-GWAS analysis helped gain new insight into causal germline variants and their impact on the aetiology of prostate cancer and translate genetic variants into therapeutic and clinically meaningful results (Farashi et al. 2019).
Meta-analysis of GWASs can be performed to increase the power of association detection by analyzing more genomic variants in human and nonhuman species. For example, to discover flavor-associated single-nucleotide polymorphisms (SNPs) in tomatoes, a meta-analysis of GWASs was performed. The results of this analysis indicated that in comparison with traditional cherry tomatoes, in modern cultivation, the majority of alleles associated with high sugar levels have been lost. Such results can provide new insight into the genetics of tomato flavor and how to control it (Zhao et al. 2019a). Further, the results of GWAS meta-analysis for multiple myeloma in human has identified suggestive novel risk alleles that could better capture disease risk in individuals (Du et al. 2020).
In post-GWAS analysis, another applicable technique is cross-phenotype association analysis, which refers to cases when loci or genes have significant associations with multiple traits. One of the limitations of quantitative trait analysis approaches, such as GWAS, is the challenge of identifying epistasis and pleiotropy. Epistasis refers to the influence of genetic mutations on other mutations, and pleiotropy refers to a phenomenon in which a single locus can control multiple phenotypic characteristics. Epistasis can cause pleiotropy, and pleiotropy is known to be an underlying cause of cross-phenotype associations (Polster et al. 2016).
This review considers the challenges of detecting truly causal variants from GWAS, identifying the functions of identified loci, and understanding the contributions of most identified loci to the pathogenesis of complex traits and the subsequent application of post-GWAS analysis techniques to overcome these challenges. Then, different post-GWAS integrative and cross-phenotype association analysis methods, considering epistasis and pleiotropy, that can provide valuable information from GWAS results, are discussed. The post-GWAS study design is indicated in Fig. 1. In addition, Box 1 summarizes the objectives and types of the post-GWAS method reviewed in this paper and key post-GWAS methods, including LD score regression, genetic correlations, and polygenic risk scores (PRSs), are discussed in Box 2.

One set of study is shown in large circle. Significant SNPs detected from the GWAS results can be causative variants, shown in small area, or those in LD with causals for which functional annotations are required, shown in another area. The post-GWAS integrative analysis, shown in the next circles below others, combining GWAS results with somatic or transcriptomic data, can be used to boost the GWAS power. Such analysis and its results for each phenotype can be used in cross-phenotype associations and phenotypic comparisons, using another set of study shown in different labeled circle.
Causality detection
According to the American College of Medical Genetics and Genomics, a genetic variant is causative if it is involved in a specific phenotype development. These phenotypes can be human diseases, behaviors, morphology (e.g., height), economic success, and also behavioral, functional, and productive phenotypes in plants and animals (Richards et al., (2015)). It is possible that a pathogenic variant is not causative; this can happen when nonfunctional and benign variants are involved in the pathogenesis of the patient’s phenotype. Based on the genomic architecture, the density of genotype data, selection signals, and genotyping technologies including SNP array designs either to include SNPs inside known genes or not, GWAS can identify the causative loci or those in linkage disequilibrium (LD) with them. Common tag SNPs can identify causal variants, their variance proportion, and their true effect size to help account for the missing heritability in GWAS (Boudellioua et al. 2017). Comparing gene-phenotype associations with patient phenotypes, a method called PhenomeNET Variant Predictor (PVP) was developed; this approach considers patients’ phenotypic similarities to rank potential candidate genes and facilitate causal variant identification. PVP merely depends on the phenotypes of the modeled organism. It is only applicable when variants are in known disease genes, and it does not provide information regarding oligogenic or digenic inheritance. DeepPvP (Boudellioua et al. 2019) and OligoPVP (Boudellioua et al. 2018) are developed methods that can be employed for PVP, which is useful for causal variant detection of complex traits. The performance of PVP has been evaluated by the previous study on congenital Hypothyroidism (CH) for potentially pathological variant detection, which analyzes a series of exomes in the UK10K dataset when the results indicate likely causative variants (Boudellioua et al. 2017).
To examine the effect of causal variants on a specific disease, Mendelian randomization (MR) is a suitable approach. The name MR comes from Mendel’s law of independent assortment when an individual’s genotype is formed randomly during segregation (Grover et al. 2017). In recent years, diverse MR methodologies have been developed, and the selection of an appropriate method depends on considering a combination of conditions such as data availability, number of SNPs, and correlations between SNPs. For example, two-stage least squares, limited information maximum likelihood, inverse variance weighted, MR-Egger, weighted median regression, multivariable MR, Bayesian MR, structural mean models, and generalized methods of moments are different MR strategies that provide causal estimates for genetic instruments (Kou et al. 2020). There are also some R packages, such as TwoSampleMR and PathD, and a STATA package called MRrobust for MR analysis (Davis et al. 2018). One previous study has employed MR in osteoporosis for causal variant inference and potential risk factor detection. Horizontal pleiotropy, LD, population stratification, trait heterogeneity, the complexity of association, dynastic effects, clinical period effects, selection bias, and weak instrument bias are some limitations of MR that can make it more complicated (Kou et al. 2020).
On the other hand, propensity score approaches are conditional probability assignments that can be applied in population-based genetic association studies to obtain valid estimates and address confounders such as disease and patient characteristics or genetic ancestry. For example, the combination of principal components and propensity scores (PCAPS) can be used to address confounders due to population stratification. The advantage of using PCAPS is the ability to detect true associations and reduce false-positive findings in GWASs by capturing and summarizing the variability in principal component analysis. PC can be carried out on GWAS results using EIGENSOFT software, whose predictions in the logistic model can be employed in PCAPS estimation. Compared to other PCA methods, PCAPS can correctly identify false-positive results. PCAPS has been examined as a practical and innovative way for testicular cancer to correct GWAS population stratifications and false-positive identification (Zhao et al. 2018). One of the other developed methods is the propensity score adjustment method (PSAM), which uses estimated propensity scores to adjust for the influences of epistasis or correlations. This method tests for single locus associations and uses genetic variant interactions or correlations to adjust for their effects and account for the missing heritability. The PSAM methodology starts with SNP subset selection and estimation of propensity scores and disease associations for each SNP. Next, univariate logistic regression is used for each SNP, and stepwise multivariate logistic regression is performed using the logit model. Without increasing the model complexity, the PSAM can increase the power of logistic regression tests for single-point association analysis when accounting for factors such as missing heritability. PSAM can be employed to determine treatment and outcome association. Furthermore, some treatment and outcome spurious associations caused by covariant confounders can be removed using PSAM. Seven simulated data types were used to evaluate the PSAM performance, and the result indicated a 15% improvement in the power of disease association identification compared to other methods. Afterward, the results of performing PSAM for rheumatoid arthritis and immunity have identified significant associated SNPs (Rai et al. 2018). One limitation of propensity score approaches is that they do not ensure balance in unmeasured and confounders and cannot substitute for randomization. The methods that are mentioned for causality detection, along with their applications and limitations, are explained in Table 1.
Functional annotations
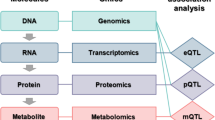
In the identification of disease-associated genetic variants, although the GWAS method is powerful, it cannot directly address genetic association signals, which are a set of variants within a locus that can influence target genes and are associated with a complex trait (Cannon and Mohlke 2018). To address such problems, post-GWAS analysis is performed by predicting the genes identified from reported GWAS variants that are most likely to be associated with the disease (Gallagher and Chen-Plotkin 2018). The post-GWAS analysis can use eQTL (Box 1), genetic and ontology data and co-functional gene networks to predict disease-related genes. The post-GWAS analysis can also consider associations between promoters and regulatory elements to predict disease-related genes distal or proximal to regulatory elements or GWAS signals. Such post-GWAS analysis can identify disease genes and then score such variants to prioritize the most likely signals (Broekema et al. 2020). For example, in a case study of Alzheimer’s disease, post-GWAS analysis identified 131 highly scored putative risk genes among 552 candidate genes (Lin et al. 2018). Furthermore, pathway analysis and Gene Ontology (GO) (The Gene Ontology Consortium 2019) terms, mammalian phenotypes (Weng and Liao 2010), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Kanehisa et al. 2017) can be combined to analyze such results and identify the most likely candidate genes. Post-GWAS pathway analysis has been successfully employed to identify novel risk pathways and biological mechanisms of type 2 diabetes (Liu et al. 2017).
On the other hand, fine-mapping approaches can be applied for variants, which are usually combinations of functional annotations and statistics. Such studies usually include genotyping arrays for studying specific SNPs, statistical approaches for the detection of causal SNPs, and functional annotations (Osgood and Knight 2018). Usually, genes close to GWAS-identified SNPs are assumed to be high-risk genes, and distant genes are ignored. For instance, integrated post-GWAS analysis of schizophrenia has been performed to address such problems and identify distant disease risk genes by regulatory elements (Lin et al., (2016).
Within a locus, independent association signals can be determined using fine-mapping approaches that involve stepwise conditional analysis involving targeted re-sequencing (Salomon et al. 2016) and imputation (Howie et al. 2012). Then, a creditable set can be defined using a posterior probability with a Bayesian approach. Next, the functional annotations of the creditable set can be determined using National Institute of Health (NIH) roadmap studies (Romanoski et al. 2015) or the Encyclopedia of DNA Elements (ENCODE) (ENCODE project consortium 2012). For example, in type 1 diabetes, 50 susceptibility loci were examined using a Bayesian fine-mapping approach (Onengut-Gumuscu et al. 2015); For instance, a Bayesian approach has been successfully used to detect significant loci associated with 22 traits in the Kaiser cohort (Majumdar et al. 2018).
Overall, the value of the generated data is strongly related to the selected tissue or cell type. Genome editing based on clustered regulatory interspaced short palindromic repeats (CRISPR) is another approach for identifying causal variants by introducing deletion/insertion mutations in a locus (Cong et al. 2013). For example, this approach was successfully applied in a study on Parkinson’s disease (Soldner et al. 2016). In addition, a GWAS-identified locus can be edited to match orthologues of other loci. In such approaches, to identify important gene regulatory regions, genome editing can make precise changes, such as SNP mutations, to identify important gene regulatory regions (Bauer et al. 2013).
Usually, causal variants coincide with regions associated with transcription factor (TF) binding sites of chromatin interactions or histone modification and open chromatin (Rivandi et al. 2018). Data on the locations of DNA methylation, open chromatin, histone modification, TF binding sites, DNA expression and other regulatory features are publicly available from ENCODE (https://www.encodeproject.org/) (ENCODE Project Consortium 2012), the NIH roadmap epigenomics project (Zhou et al. 2015), the FunctiSNP R package (http://www.bioconductor.org/packages/release/bioc/html/FunciSNP.html) (R Core Team 2012), RegulomeDB (http://www.regulomedb.org/) (Boyle et al. 2012) and HaploReg (http://archive.broadinstitute.org/mammals/haploreg/haploreg.php) (Ward and Kellis 2011). Inferring the mechanism of causal variants is complicated because GWAS-identified loci may regulate multiple RNAs or target genes. To address this challenge, information about gene expression and chromatin interaction, regulatory data and bioinformatics developments can be useful (Rivandi et al. 2018).
Enrichr (http://amp.pharm.mssm.edu/Enrichr) is a comprehensive resource containing a collection of gene sets and their biological knowledge to further analyze GWAS results. The number of annotated gene sets in Enrichr is more than 180,184 (Kuleshov et al. 2016). Moreover, another web-based platform that can be used for GWAS results’ functional annotation and genetic causal variants’ prioritization is FUMA, http://fuma.ctglab.nl. It provides adequate insight into the genetic variants’ biological implications by combining biological data repositories and tools (Watanabe et al., (2017)). In addition, being training on eQTL fine-mapping, Expression Modifier Score (EMS) is a genomic score method used to predict regulatory effects of variants on gene expression and could leverage epigenetic marker prediction. Among other genomic score methods, the EMS has higher prediction accuracy and is useful for regulatory variant prioritization. Initially, score bins were predicted for that method, and then the fraction was calculated for positively labeled samples to scale the output score and derive EMS. EMS has been validated and used preferably to QTLs statistical fine-mapping. Then using the UK Biobank (UKBB) dataset (Bycroft et al. 2018) for hematopoietic traits, the Finucane lab, https://www.finucanelab.org/, used the EMS to prioritize putative causal variants of non-coding regions (Wang et al. 2021).
One of the most important challenges in the field of GWAS is that most significant SNPs identified through GWASs fall outside of coding regions; thus, the function and contribution of most loci to the pathogenesis of complex diseases are largely unknown (Mortezaei et al. 2017). Thus, it is critical to understand the biological functions, roles and disease effects of genetic variants. The detection of functional genetic variants in non-coding elements is discussed in Box 3.
Post-GWAS integrative analysis
GWAS results can be compared with prior findings to get more valuable genetic results which can be used for real-world medical applications. For GWAS summary-level data, a comprehensive collection can be assessed using the GWAS Central database (Beck, Shorter T (2019)) or GWAS database (GWASdb) (http://jjwanglab.org/gwasdb) (Lin et al., (2016)) to obtain access to unified and combined data. GWAS Central is a collection of metadata and GWAS summary-level data from many sources, including the Open Access Database of Genome-wide Association Results (Johnson and O’Donnell 2009) and the National Human Genome Research Institute-European Bioinformatics Institute (NHGRI-EBI) (Buniello et al. 2019), published or unpublished GWAS data, etc. One of the advantages of using GWAS Central is that all available summary-level data in that database are available for use, rather than limiting the data to only results with significant p-values (Beck, Shorter T (2019)). In comparison with GWAS Central, in GWASdb, there is a larger number of GWAS publications that studied population-specific traits (Lin et al., (2016)).
Integrating GWAS results with other resources, such as clinical findings, co-functional genes, somatic mutations, metabolite-transcript correlation, and eQTL data, can provide valuable information about the genetics of quantitative traits (Wang et al. 2016). For example, network-based integrative analysis of GWAS biological signals with networks of co-functional genes provided an opportunity to augment GWAS findings and detect highly probable candidate genes in association with quantitative traits in Arabidopsis thaliana (Lee and Lee 2018). Integrative analysis has also been applied to combine GWAS findings with a network of metabolite-transcript correlations for Arabidopsis. This strategy can be used to identify gene-metabolite associations and discover novel genes in relation to the metabolites (Wu et al. 2016). Applying large-scale integrative analysis of GWAS data with methylation QTLs could also identify multiple disease-specific genes and pathways and provide novel insight into their genetic mechanisms (Zhao et al. 2017). In addition, the integration of GWAS results with epigenomic data can be achieved by applying the GWAS3D database for the identification of genetic variants with the ability to affect regulatory elements such as enhancers and promoters. Evaluation of GWAS3D was successfully performed for plasma low-density lipoprotein cholesterol to prioritize regulatory variants (Li et al. 2013).
It has been shown that integrative analysis that links genetic variations with their biological roles and functions is important and useful in genetic prediction. Integrative analysis of GWAS results has revealed some genes in association with obesity-related phenotypes, considering their contribution to the regional fat distribution (Ahn et al. 2019). In addition, in previous studies, some hub genes in relation to milk yield in Mediterranean buffaloes were found using co-expression network analysis and GWAS data (Deng et al. 2019).
Germline variants and somatic mutations
Single-cell analysis can e.g., be employed for studies of cancer, as a disease caused by uncontrolled invasiveness and proliferation and somatic mutations (Ren et al. 2018). For instance, somatic single nucleotide variants on bone marrow were discovered performing enhanced whole-genome sequencing (Petti et al. 2019). In addition, somatic mutations can be caused by mosaic chromosomal alterations of specific tissues using genome re-sequencing or array genotyping data. As an illustration, it has been previously identified that the risk of elevated hematological cancer is ten times more in individuals with mosaicism chromosomal alterations (Loh et al. 2018). Another study employed SNP-array data from UK Biobank to detect mosaic chromosomal alterations of blood cancer (Loh et al. 2020).
For an analysis of prostate cancer, 305 individuals with aggressive tumors and 52 control samples were selected from the Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research Network et al. 2013). In addition, 61 germline variants in association with prostate cancer were downloaded from the GWAS catalogue database (Welter et al. 2014), and information about somatic mutations was obtained from Catalogue of Somatic Mutations in Cancers (COSMIC) (Tate et al. 2018). Then, possible genetic cooperation and oncogenic interactions between germline variants and somatic mutations were investigated. Then, for enrichment analysis of germline and somatic mutations, IPA can be used (Kramer et al. 2014). The results highlighted the power of post-GWAS integrative analysis to determine the biological context of aggressive prostate cancer (Mamidi et al. 2019). Another study by Wu et al. (2019) integrated germline and somatic mutations for carcinogenesis-related gene identification in triple-negative breast cancer. As a result, 237 genes were discovered that were functionally related to germline and somatic mutations. These functionally related germline and somatic mutations can be used for prognostic marker identification and the development of prevention strategies (Wu et al. 2019).
In addition, using gene expression data, germline and somatic mutation information has been integrated, and 124 common genes associated with prostate cancer have been identified. In this study, to gain insight into the biological function of germline and somatic mutations, molecular networks of differentially expressed genes were generated and biological pathway analyses were performed using IPA (Kramer et al. 2014). The results of such analyses can be used to discover interactions between germline and somatic mutations and the putative functional bridges between them (Mamidi et al. 2019). In addition, the results of such studies can demonstrate that the somatic evolution of tumors can be affected by germline variants. The existence of germline and somatic mutation interactions can indicate the existence of some cooperation between such mutations, although the mechanism of such interactions has not been investigated, and more research is required (Jia and Zhao 2016).
Relations among germline variants, somatic mutations, and genetic drug targets of complex human disorders can be employed to provide new insights into complex human diseases. The genetic findings of such studies can be translated into clinical applications (Chen et al. 2019). For example, such integrative analyses of the genetics of cancer (Ung et al. 2016) and the genetics of neurodegenerative diseases (NDs) (Mortezaei et al. 2019) have been reported. These studies can help identify genetic modules with clinical roles in the initiation, development, and treatment of complex human disorders, such as cancer or NDs.
In such studies, a directed functional interactome, node classes of germline variants, somatic mutations and drug targets for complex human diseases were combined, and the relative positions of the node classes were identified by network analyses. For the created node classes containing germline variants, somatic mutations and drug targets, the genetic functional interactions were downloaded from the Reactome database (http://www.reactome.org/pages/download-data/) (Jassal et al. 2019). As indicated in Fig. 2, through integration of germline variants, somatic mutations, and drug targets via network-based analysis, the hierarchical structure of the networks was also evaluated to compare the roles and importance of elements in those biological networks. As a result, all such studies revealed that drug targets are the most important factors functionally influencing others, followed by somatic mutations and germline variants (Mortezaei et al. 2019; Ung et al. 2016).

A network of germline variants from GWAS results, with networks of somatic mutations and genetic drug targets, was used to create an integrated network containing germline variants, somatic mutations and genetic drug targets. Then, the results of integrative genetic analysis performed using hierarchical network analysis and network centralities were used to identify novel candidate genes important in the pathogenesis of complex diseases. As an example, the results of such an analysis used to assess the importance of gene mutations in cancer and neurodegenerative diseases in humans is shown. The integrative genetic analysis shows that somatic mutations in relation to cancer or neurodegenerative diseases in human beings have strong, independent effects on their genetic drug targets, which can be used for individual treatments.
Proteins that bound to drugs in nonhuman studies were shown to have some homologs in humans, identified as potential drug targets in humans and retrieved from the Protein Data Bank (PDB) database (wwPDB Consortium 2019). Somatic and germline mutations also occur in nonhuman species such as animals and plants. It has been demonstrated previously that the rate of somatic mutations is higher than that of germline mutations in animals such as mice, and the rates of both kinds of mutations are higher than those of mutations in humans. In plants, somatic mutations occur during mitotic cell division in gametophytes or sporophytes, and gametic mutations occur during meiosis (Milholland et al. 2017). Considering similarities or differences between genetic drug targets and germline or somatic mutations in humans and nonhuman species, such integration analysis, can be performed for all kinds of species.
GWAS and transcriptomic data
Integrating GWAS and eQTL data provided novel susceptibility genes in relation to obesity and some clues for studies of their mechanisms (Liu et al. 2018). GWAS data can also be integrated with eQTL and protein-protein interaction data to detect disease-associated genes and prioritize candidate genes. With such studies, one can go beyond GWAS, eQTL, and protein-protein interaction approaches (Wang et al. 2018). When integrating GWAS with transcriptome data on complex traits, estimation of the causal effects of gene expression can be performed by applying the MR approach. Transcriptome-wide Mendelian randomization (TWMR) is a multivariable MR approach integrating GWAS summary-level data with eQTLs to estimate the causal effects of gene expression on complex traits. Previously, TWMR has been successfully applied to assess gene expression’s causal associations with 43 complex traits (Porcu et al. 2019).
A previously conducted study analyzed GWAS results using regulatory datasets, such as eQTL, to identify causal variants (Lin et al. 2018). Another study concentrated on integrating GWAS results with eQTL data for disease gene identification, and then the strength of candidate genes was scored using ontology datasets (Peat et al. 2020). Another study combined GWAS results with eQTL data to identify Alzheimer’s disease-associated genes and prioritize significant SNPs (Zhao et al. 2019b). Functional enrichment analysis of SNPs and eQTL-based SNP ontology platforms have been constructed before, which is helpful to identify significant SNPs in association with complex diseases, such as neurodegenerative disease (Li et al. 2016).
From the genotype-tissue expression (GTEx) consortium, eQTL data were downloaded and integrated with GWAS summary statistics for body mass index to identify signals with the same causal variants. The results of such analyses are tissue specific, which indicates that different tissues and molecular mechanisms are involved (The GTEx Consortium 2013). Another study integrated GWAS data with eQTL data from 44 tissues selected from the GTEx project. In that study, regulatory variations were used to assess GWAS tissue specificity and to discover causal genes in multiple tissues. Several approaches have been used for such integrative analysis to identify genetic variations of different diseases. These approaches include heritability analysis, enrichment analysis, linking contributions of tissue-specific eQTLs, and true positive rate estimation (Mortlock et al. 2020). Then, in such studies and for all tested GWAS tissues, Bonferroni correction was used to assess significant GWAS-trait pairs. Finally, a gene-set enrichment analysis was used to test for GWAS and eQTL target genes. For instance, to identify potential risk alleles and causal genes, eQTL has been applied on gene expression, genotype data from the GTEx project for colon tissue, and TCGA data for colorectal tumor tissue (Loo et al. 2017).
Cross-phenotype associations
It has also been demonstrated that detected loci or genes can have significant associations with multiple traits, referred to as cross-phenotype associations (Li et al. 2017). Some cross-phenotype association tests can be used to boost the power of GWASs. For example, multiple GWASs have demonstrated associations of a gene desert on chromosome 8p24 with chronic lymphocytic leukemia and colon, breast, prostate, bladder and ovarian cancers (Turnbull et al. 2010). Other studies have demonstrated that a functional variation of the PTPN22 gene is associated with systemic lupus erythematosus, type 1 diabetes, Graves’ disease and rheumatoid arthritis (Solovieff et al. 2013).
Statistical methods for detecting cross-phenotype associations have been broadly classified into univariate and multivariate analyses (Box 4) (Broadaway et al. 2016). For example, multivariate analysis of variance (MANOVA) can be used in regression of multiple phenotype analysis (Yang et al. 2019). One of the methods that can be applied in this direction is Fisher’s combined P value method (Li and Zhu 2017). In addition, a linear mixed model can be used for multivariate analysis. Table 2 summarizes some properties, including the advantages/disadvantages of using univariate vs. multivariate approaches (Saccenti et al. 2014). In cases where the phenotypes are non-normally distributed or categorical, cross-phenotype analysis can be performed using modifications of original regression, generalized estimating equations and the Bayesian framework (O’Reilly et al. 2012). There exist some multi-phenotype methods, such as MV-BIMBAM software (Shim et al. 2015) for multivariate association analysis, which uses a Bayesian model comparison, and multivariate-linear mixed model (MV-LMM), which can be used for related phenotypes. For MV-LMM, two types of optimization algorithms can be used: an expectation-maximization (EM) algorithm followed by a Newton-Raphson (NR) algorithm, which combines the stability of EM with the faster convergence of NR (Zhou and Stephens 2014). As an illustration, multivariate, univariate, and bivariate analyses have been successfully performed on a European population of 43,870 cardiovascular and neurological diseases (Zhang et al. 2019).
The phenome-wide association study (PheWAS) (Box 4) approach can be used to investigate cross-phenotype associations, to demonstrate genetic architecture in relation to genes and pleiotropy (when one locus affects more than one trait or phenotype) and for diagnosis (Bush et al. 2016). In network analyses (Box 4), SNP-SNP interaction networks from GWASs have been constructed graphically, and the BridGE approach (Fang et al. 2017) was applied to identify single or multiple biological pathways enriched for that interaction; For example, SNP-SNP interaction analysis has been performed for cardiovascular risk in autoimmune diseases, which helped classify them into more associated groups (Perrotti et al. 2017).When BridGE searches for within/between or hub pathway models, the results of such pathway-level interaction analysis can provide useful information about increasing or decreasing the risk of related diseases (Wang et al. 2017); For instance, significant interactions have been identified between type 2 diabetes, Parkinson’s disease, breast cancer, prostate cancer, and schizophrenia by widely applying BridGE successfully (Fang et al. 2019). There are some limitations in this kind of network analysis. These limitations include phenotypic differences across studies and limited individual-level phenotype or genotype information from networks of summary statistics. These limitations were addressed in a study by Verma et al. (2019) in which a single-source, electronic health record (EHR) (Agrawal and Prabakaran 2020) was used for specific definitions of phenotypes and an individual genotyping platform was applied. Finally, the results of 31,017 PheWASs were used to create a disease-disease network (DDN). For example, DDNs have been successfully constructed to identify genetic similarities between diseases, such as rheumatoid arthritis, type 1 diabetes, and multiple sclerosis (Verma et al. 2019).
Electronic health record (EHR)-based PheWASs can be used to identify cross-phenotype associations, construct DDNs on the basis of shared associations and understand genetic similarities between diseases. An EHR-based comprehensive PheWAS has been performed to provide the landscape of associations across diseases and quantitative traits (Verma et al. 2018). The results of such analyses revealed previously reported associations between type 1 diabetes, morbid obesity and a primary hypercoagulable state (Wellcome Trust Case Control Consortium 2007). In addition, a large number of genetic variants indicated strong connections between autoimmune disorders such as type 1 diabetes, psoriasis, rheumatoid arthritis and multiple sclerosis. This indicates that even if different types of tissues are affected in each autoimmune disorder, they all share similar genetic components via shared genetic pathways (Tettey et al. 2015).
DDNs are bipartite networks that can be constructed and visualized using Gephi software (https://gephi.org), in which statistical packages can be used as plug-ins for network analysis. In the analysis of DDNs, one of the key goals is the identification of strongly linked diseases within and between disease classes and the identification of meaningful connections. In addition, integrating genetic functional knowledge with association results can broaden our understanding of biologically relevant findings (Halu et al. 2019). Then, DDNs and epigenetic knowledge can be integrated to examine tissue-specific changes (Verma et al. 2019).
Network analysis, such as community detection, can also be applied to extract subnetworks of diseases that are biologically relevant. There are various community-detection techniques, such as Louvain’s method (Blondel et al. 2008), that use Gephi software (Bastian et al. 2009) for subnetwork detection. For such analysis, only SNPs in the enhancers of specific tissues were considered. As a result, the liver has been found to have the largest number of associated diseases, such as hyperlipidemia, essential hypertension, chronic non-alcoholic diseases, cardiovascular diseases, morbid obesity and cirrhosis of the liver (Verma et al. 2019). Network analysis can be extended to include associations between EHR clinical laboratory measures and genetic variants to conduct large studies based on gene-trait associations. Another study by Verma et al. (2018) used RNA-Seq data from the roadmap epigenome for genetic association periodization based on gene expression measures. This study first calculated the correlation between gene expression and chromatin state, generated a gene expression measurement matrix and finally performed regression analysis between chromatin model binary measures and gene expression. This type of analysis can be applied to improve the understanding of the effects of genetic variations on phenotypes when explorations beyond those of protein-coding regions are possible. Epigenetic knowledge helps identify associated diseases and their biological relevance in the context of cross-phenotype associations (Gonzalez-Serna et al. 2020). The cross-phenotype methods are discussed further and classified in Box 4.
Using principal component-based methods for cross-phenotype associations in flies, some regulatory loci have been identified that jointly associate with multiple metabolic pathways. In addition, the cross-phenotype association test in Drosophila was used to detect metabolism-related genes. These results can be applied for genotype-to-phenotype mapping of metabolic traits. In addition, in Drosophila, cross-phenotype association mapping has been used to examine starvation resistance, glucose content and body weight (Nelson et al. 2016). This study applied phenotype measurements from the Drosophila Genetic Reference Panel and the SMAT R package (Schifano et al. 2013) for the given traits. These analyses revealed significant associations between triglyceride levels, starvation resistance and CG7560 and cht12 loci. In cross-phenotype tests using enriched association signals, starvation resistance revealed associations with genes enriched in ventral cord development and glucose content. These results illustrated that characters affecting the central nervous system are associated with hyperactivity during starvation.
On the one hand, the Enhancing Neuroimaging Genetics through Meta-analysis (ENIGMA) consortium started more than ten years ago, aiming to study neuroimaging genetics on a large scale. Such analysis used more than 50,000 individuals to indicate robustly associated genetic markers with brain function and structure. The analysis results were identified in more than 200 loci having significant association with brain variations. Afterward, ENIGMA applied multivariate methods to fulfill quantifying challenges in brain networks’ complex relationships. In addition, the cross-disorder groups of ENIGMA used multiple genomic data to answer transdiagnostic questions. For this group, an exemplar approach is examining brain organization for psychiatric disorder patients with first-degree unaffected relatives (Thompson et al. 2020).
Epistasis and pleiotropy
Epistasis refers to a case in which genetic mutations are influenced by the presence or absence of other genetic mutations. Therefore, the expression of genes in a locus is altered by another locus. In such a case, at different loci, multiple genes interact with each other to affect a trait. Epistasis effects are known to be one of the factors underlying missing heritability. This is because epistasis can reveal genetic interactions and provide insights for complex genotype and phenotype mapping that cannot be achieved from association studies. Within gene regulatory networks and biological pathways, the result of physical interactions between biomolecules is called biological epistasis. On the other hand, genotype and phenotype relationships summarized using mathematical modeling and their deviations from additivity are called statistical epistasis. Therefore, biological epistasis and statistical epistasis provide two different perspectives and are consistent with strategies such as gene-gene, SNP-SNP, and protein-protein interactions (Slim et al. 2018).
On the other hand, pleiotropy is a phenomenon where a single locus influences or controls multiple phenotypic characteristics. The pleiotropy is known as an underlying cause of cross-phenotype associations. In other words, cross-phenotype associations are more general than the pleiotropy capable of occurring in biological pleiotropy, phenotypic causal relationships, spurious associations, study design, and confounder biases. In genetic epidemiologic studies, cross-phenotype associations are often incorrectly interpreted as pleiotropy examples, while pleiotropy is the only possible cross-phenotype association explanation. Therefore, a careful dissection of cross-phenotype associations is necessary for the detection of true pleiotropic loci. Both mentioned univariate and multivariate methods in the “Cross-phenotype association” section are pleiotropy informed considering cross-phenotype correlations (Salinas et al. 2017).
The ubiquity and the relation of pleiotropy with human effect size can be examined using the GWAS catalog as a comprehensive database (Welter et al. 2014). It has been found that nearly half of the genes in that database are associated with more than one disease, and that number will continue to increase. In addition, in the UniProt database, ~12% of protein-coding genes were identified as pleiotropic (The UniProt Consortium 2018). In the case of pleiotropy, in which a gene is associated with more than one phenotype, the genes are likely to be involved in many biological processes and to have a strong phenotypic effect. In quantitative trait analyses such as GWASs, one of the limitations is the inability to detect epistasis and pleiotropy (Polster et al. 2016).
Since pleiotropy depends on genetic interactions, epistasis can cause pleiotropic variation at a locus. This is because the way genes affect more than one trait depends on their interactions with other genes. Therefore, for the evolution of pleiotropy, genetic variations caused by epistasis are necessary. In all types of organisms, such as plants, viruses, bacteria, and humans, previous studies have identified epistasis and pleiotropy. For example, it has been identified that in human immunodeficiency virus (HIV) infection and multiple drug resistance, epistasis and pleiotropy play fundamental roles (Polster et al. 2016). The pleiotropy that explains genetic variants contributing to multiple traits is known as an underlying cause of cross-phenotype associations. In other words, cross-phenotype associations are more general than the pleiotropy that can occur in biological pleiotropy, phenotypic causal relationships and spurious associations. Both the univariate and multivariate methods mentioned in the “Cross-phenotype association” section are pleiotropy informed considering cross-phenotype correlations (Salinas et al. 2017).
Modeling of discoveries from GWASs can be performed using effect direction meta-analysis (EDME) for pleiotropy quantification. EDME has been applied in cattle to discover trait variation and better understand the biology of complex traits. In that study, EDME of GWASs on cows and dairy bulls was performed to discover pleiotropic variants, their related affects and the biology behind each complex trait (Xiang et al. 2020).
Phenotypic comparisons
Following the GWAS and cross-phenotype association analysis approach, a study by Gu et al. (2019) concentrated on genetic similarities between phenotypes, employing a statistical approach that is analogous to Fisher’s probability test for a set of SNPs relating to different molecular traits. Based on this approach, the similarities between phenotypes can be determined using functionally related genes and their common molecular mechanisms. As an example, in that study, it was found that breast cancer, prostate cancer, lung cancer, fasting glucose and fasting insulin were clustered together. It has also been determined that common molecular underpinnings, such as AMP/GMP signaling, insulin/NAPDH oxidase/ROS and apoptosis, play important roles in connecting the mentioned phenotypes (Gu et al. 2019).
GWAS summary-level data is an informative source of pooled data that can be used to compare phenotypes within and between species. For example, GWAS Central (Beck, Shorter T (2019)) allows unified data visualization and interrogation by restricting the display of risk alleles, and GWASdb (Lin et al., (2016)) integrates comprehensive resources for data content extension and population studies. The inclusion of additional ontologies such as those in the systematized nomenclature of medicine clinical terms (SNOMED CT) is an example of the extension of information about semantic phenotypes that can be applied for future planning (Al-Hablani 2017). In addition, the reverse GWAS (RGWAS) approach uses the genetic basis of multiple traits from GWAS results to classify phenotypes and produce homogeneous subtypes of samples. RGWAS has two steps: the initial step includes multitrait GWAS dataset clustering with the regression method, and the second step is biological assessment between those clusters. The “rgwas” R package (https://github.com/andywdahl/rgwas) is available for RGWAS implementation. RGWASs can handle residual trait correlations, covariates, quantitative traits and mixed binary data. Propagating first-step uncertainty is not possible using the RGWAS approach, one of the limitations of this approach that needs to be considered in the future. For example, RGWAS has been successfully applied to recover subtypes of stress to depressive disorder and identify metabolic traits (Dahl et al. 2019).
To understand the biology of diseases, their prognosis and their treatment, it may be essential to distinguish their subtypes. For example, different subtypes of breast cancer have been distinguished by prognoses, population structure, treatment responses, and different genetic risk factors (Iqbal et al. 2015). Gene-environment interactions (Fairfax and Knight 2014), disease misclassification (The Brainstorm Consortium 2018), and gene-gene interactions (Fang et al. 2019) create distinct subtypes of complex diseases.
Phenotypic comparisons can also be performed via post-GWAS integrative analysis. For example, as shown in Box 5, integrating germline variants from GWASs with somatic mutations revealed similarities between cancer and NDs.
Conclusions
In the last decade, the number of established SNP-trait associations from GWASs has increased dramatically, but the determination of causal variants from them remains a major challenge. Post-GWAS analysis can be used to address such challenges in the detection of causal variants from GWASs and to determine their mechanisms of action. On the other hand, it is critical to understand the biological functions of genetic variants and how they can affect diseases. This requires interpreting the functions and contributions of most loci to the pathogenesis of complex diseases, considering the fact that most significant SNPs identified through GWASs fall outside of coding regions. Additionally, to identify functional genetic variants, GWAS results can be combined with chromatin features, genome-wide maps and genetic transcription data to help overcome challenges in the field.
In addition, integrative analysis of GWAS results with co-functional genes, clinical findings, eQTL data and metabolite-transcript correlations can provide valuable information about the genetics of complex diseases, and such results can be translated into clinical applications. For example, post-GWAS integrative analysis has revealed that the somatic evolution of tumors can be affected by germline variants. Such interactions between germline variants and somatic mutations can result from cooperation between them. This interaction mechanism has not been investigated, and further research is required to answer this question.
On the other hand, the challenges of detecting epistasis and pleiotropy in quantitative analysis approaches, such as GWASs, can be overcome using post-GWAS analysis. The pleiotropy explaining the genetic variants that contribute to multiple traits is an underlying cause of cross-phenotype associations. It has also been concluded that in GWASs, focusing on one trait can result in missing the opportunity to evaluate multiple phenotypes, especially when cross-phenotype associations exist; in such cases, phenome-wide data can be used to improve the statistical power of genetic association studies. Similarities between phenotypes can be measured from GWAS summary-level data. The results of such analyses followed by enrichment analysis can facilitate the development of effective treatment and prevention options for complex diseases. For example, GWAS results were used to compare human cancer with NDs, which indicated that the genetic drug targets for both kinds of diseases were responsible for initiating signaling cascades. Another common conclusion from analyses of GWAS results for both cancer and NDs is that drug targets and somatic mutations correspond to bottleneck proteins that can transfer signals to the nucleus. Such a conclusion can be used to develop a framework for future studies and to better understand the genetics of different complex diseases. The review also discussed and provided some examples that post-GWAS methods, which can be used to weight the results, can be performed for humans or for nonhuman species.
References
Agrawal R, Prabakaran S (2020) Big data in digital healthcare: lessons learnt and recommendations for general practice. Heredity 124:525–534
Ahn J, Wu H, Lee K (2019) Integrative Analysis Revealing Human Adipose-Specific Genes and Consolidating Obesity Loci. Sci Rep 9(1):3087
Al-Hablani B (2017) The Use of Automated SNOMED CT Clinical Coding in Clinical Decision Support Systems for Preventive Care. Perspect Health Inf Manag 14 (Winter): 1-8.
Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, Brainstorm Consortium. et al. (2018) Analysis of shared heritability in common disorders of the brain. Science 360:1313
Atkinson EG, Maihofer AX, Kanai M, Martin AR, Karczewski KJ, Santoro ML et al. (2021) Tractor uses local ancestry to enable the inclusion of admixed individuals in GWAS and to boost power. Nat Genet 53(2):195–204
Bastian M, Heymann S, Jacomy M (2009) Gephi: An Open Source Software for Exploring and Manipulating Networks. ICWSM. https://vbn.aau.dk.
Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C et al. (2013) An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 342(6155):253–257
Beck T, Shorter T, Brookes AJ (2019) GWAS Central: a comprehensive resource for the discovery and comparison of genotype and phenotype data from genome-wide association studies. Nucleic Acids Res 98(D1):D933–D940
Blondel VD, Guillaume JL, Lambiotte R, Lefebvre E (2008) Fast unfolding of communities in large networks. J Stat Mech Theory Exp 2008:P10008
Boudellioua I, Razali RBM, Kulmanov M, Hashish Y, Bajic VB, Goncalves-Serra E et al. (2017) Semantic prioritization of novel causative genomic variants. PLoS Comput Biol 13(4):e1005500
Boudellioua I, Kulmanov M, Schofield PN, Gkoutos GV, Hoehndorf R (2018) OligoPVP: Phenotype-driven analysis of individual genomic information to prioritize oligogenic disease variants. Sci Rep 8:14681
Boudellioua I, Kulmanov M, Schofield PN, Gkoutos GV, Hoehndorf R (2019) DeepPVP: phenotype-based prioritization of causative variants using deep learning. BMC Bioinform 20:65
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M et al. (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22(9):1790–1797
Broadaway KA, Cutler DJ, Duncan R, Moore JL, Ware EB, Jhun MA et al. (2016) A Statistical Approach for Testing Cross-Phenotype Effects of Rare Variants. Am J Hum Genet 98(3):525–540
Broekema RV, Bakker OB, Jonkers IH (2020) A practical view of fine-mapping and gene prioritization in the post-genome-wide association era. Open Biol 10:190221
Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics Consortium. et al. (2015) LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47(3):291–295
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C et al. (2019) The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47:D1005–D1012
Bush WS, Oetjens MT, Crawford DC (2016) Unravelling the human genome-phenome relationship using phenome-wide association studies. Nat Rev Genet 17(3):129–145
Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K et al. (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature 562:203–209
Cannon ME, Mohlke KL (2018) Deciphering the Emerging Complexities of Molecular Mechanisms at GWAS loci. AJHG 103(5):637–653
Chen HT, Chin YM, Low S-K (2019) The Roles of Common Variation and Somatic Mutation in Cancer Pharmacogenomics. Oncol Ther 7:1–32
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N et al. (2013) Multiple Genome Engineering Using CRISPR Cas System. Science 339(6121):819–823
Dahl A, Cai N, Ko A, Laakso M, Pajukanta P, Flint J et al. (2019) Reverse GWAS: using genetics to identify and model phenotypic subtypes. PLoS Genet 15(4):e1008009
Davis NM, Holmes MV, Smith CD (2018) Reading Mendelian randomization studies: a guide, glossary, and checklist for clinicians. BMJ 362:K601
Deng T, Liang A, Liang S, Ma X, Lu X, Duan A et al. (2019) Integrative Analysis of Transcriptome and GWAS Data to identify the Hub Genes Associated with Milk Yield Trait in Buffalo. Fron Genet 10:36
Driver JA (2014) Inverse association between cancer and neurodegenerative disease: review of the epidemiologic and biological evidence. Biogerontology 15(6):547–57.
Du Z, Weinhold N, Song GC, Rand KA, Van Den Berg DJ, Hwang AE et al. (2020) A meta-analysis of genome-wide association studies of multiple myeloma among men and women of African ancestry. Blood Adv 4(1):181–190
ENCODE project consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414):57–74
Fairfax BP, Knight JC (2014) Genetics of gene expression in immunity to infection. Curr Opin Immunol 30C(1):63–71
Fang G, Wang W, Paunic V, Heydari H, Costanzo M, Liu X et al. (2019) Discovering genetic interactions bridging pathways in genome-wide association studies. Nat Commun 10(4274):1–18
Farashi S, Kryza T, Clements J, Batra J (2019) Post-GWAS in prostate cancer: from genetic association to biological contribution. Nat Rev Cancer 19(1):46–59
Freedman DM, Wu J, Chen H, Kuncl RW, Enewold LR, Engels EA et al. (2016) Association between cancer and alzheimer’s disease in a U.S. Medicare population. Cancer Med 5(10):2965–2976
Gallagher MD, Chen-Plotkin AS (2018) The Post-GWAS Era: from Association to Function. Am J Hum Genet 102(5):717–730
Giral H, Landmesser U, Kratzer A (2018) Into the wild: GWAS Exploration of Non-coding RNAs. Front Cardiovas Med 5:181
Gonzalez-Serna D, Ochoa E, Lopez-Isac E, Julia A, Degenhardt F, Ortego-Centeno N et al. (2020) A cross-disease meta-GWAS identifies four new susceptibility loci shared between systemic sclerosis and Crohn’s disease. Sci Rep 10(1862):1–11
Grover S, Del Greco MF, Stein CM, Ziegler A (2017) Mendelian Randomization. Methods Mol Biol 1666:581–628
Gu J, Fuller C, Zheng J, Li H (2019) An integrative analysis of GWAS and intermediate molecular trait data reveals common molecular mechanisms supporting genetic similarity between seemingly unrelated complex traits. bioRXIv: 1–30
Guo H, Li JJ, Lu Q, Hou L (2021) Detecting local genetic correlations with scan statistics. Nat Commun 12(1):2033
Halu A, De Domenico M, Arenas A, Sharma A (2019) The multiplex network of human diseases. Syst Biol Appl 5(15):1–12
Horlings HM, Shah SP, Huntsman DG (2015) Using Somatic Mutations to Guide Treatment Decisions Context Matters. JAMA Oncol 1(3):5–6
Hou L, Zhao H (2013) A review of post-GWAS prioritization approaches. Front Genet 4(280):1–6
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 44(8):955–959
Iqbal J, Ginsburg OM, Rochon PA, Sun P, Narod SA (2015) Differences in breast cancer stage at diagnosis and cancer-specific survival by race and ethnicity in the United States. JAMA 313(2):165–173
Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A et al. (2019) The reactome pathway knowledgebase. Nucleic Acids Res 48(D1):D498–D503
Jia P, Zhao Z (2016) Impacts of somatic mutations on gene expression: an association perspective. Brief Bioinform 18(3):1–13.
Johnson AD, O’Donnell CJ (2009) An Open Access Database of Genome-wide Association Results. BMC Med Genet 10(6):1–17
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45(D1):D353–D361
Klus P, Girillo D, Orfila TB, Tartaglia GG (2015) Neurodegeneration and cancer: where the disorder prevails. Sci Rep 5(1):15390
Kou X, Zhou W, He Y, Ying X, Chai S, Fei T (2020) A Mendelian Randomization Analysis to Expose the Causal Effect of IL-18 on Osteoporosis Based on Genome-Wide Association Study Data. Front Bioeng Biotechnol 8(201):1–7
Kramer A, Green J, Pollard J, Tugendreich JS (2014) Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30(4):523–530
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z et al. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44(W1):W90–W97
Lee T, Lee I (2018) araGWAB: Network-based boosting of genome-wide association studies in Arabidopsis thaliana. Sci Rep 8(1):2925
Lewis CM, Vassos E (2020) Polygenic risk scores: from research tools to clinical instruments. Genome Med 12(1):44
Li MJ, Wang LY, Xia Z, Sham PC, Wang J (2013) GWAS3D: detecting human regulatory variants by integrative analysis of genome-wide associations, chromosome interactions and histone modifications. Nucleic Acids Res 41:W150–W158
Li J, Wang L, Jiang T, Wang J, Li X, Liu X et al. (2016) eSNPO: An eQTL-based SNP Ontology and SNP functional enrichment analysis platform. Sci Rep 6:30595
Li X, Zhu X (2017) Cross-Phenotype Association Analysis Using Summary Statistics from GWAS. In: Elston RC (Second edition) Methods Mol Biol 1666: p 455–467. Humana Press, US
Lin JR, Jaroslawicz D, Cai Y, Zhang Q, Wang Z, Zhang ZD (2018) PGA: post-GWAS analysis for disease gene identification. Bioinformatics. 34(10):1786–1788
Lin J-R, Cai Y, Zhang W, Nogales-Cadenas R, Zhang ZD (2016) Integrated Post-GWAS Analysis Sheds New Light on the Disease Mechanisms of Schizophrenia. Genetics 204(4):1587–1600
Lin MJ, Liu Z, Wang P, Wong MP, Nelson MR, Kocher JPA et al. (2016) GWASdb v2: an update database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res 44(D1):D869–D876
Lin J-R, Jaroslawicz D, Cai Y, Zhang Q, Wang Z, Zhang ZD (2018) PGA: post-GWAS analysis for disease gene identification. Bioinformatics 34(10):1786–1788
Liu L, Fan Q, Zhang F, Guo X, Liang X, Du Y et al. (2018) A Genomewide Integrative Analysis of GWAS and eQTLs Data Identifies Multiple Genes and Gene Sets Associated with Obesity. BioMed Res Int 2018(2):1–5
Liu Y, Zhao J, Jiang T, Yu M, Jiang G, Hu Y (2017) A pathway analysis of genome-wide association study highlights novel type 2 diabetes risk pathways. Sci Rep 7(1):12546. https://doi.org/10.1038/s41598-017-12873-8
Loh PR, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF et al. (2018) Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 559:350–355
Loh PR, Genovese G, McCarroll SA (2020) Monogenic and polygenic inheritance become instruments for clonal selection. Nature 584:136–141
Loo LWM, Lemire M, Le Marchand L (2017) In silico pathway analysis and tissue specific cis-eQTL for colorectal cancer GWAS risk variants. BMC Genom 18(1):381
Lu Q, Jin C, Sun J, Bowler R, Kechris K, Kaminski N et al. (2016) Post-GWAS Prioritization Through Data Integration Provides Novel Insights on Chronic Obstructive Pulmonary Disease. Stat Biosci. 2016:1–17
MacArthur J, Bowler E, Cerezo M, Laurent G, Hall P, Hastings E et al. (2017) The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res 45(D1):D896–D901
Majumdar A, Haldar T, Bhattacharya S, Witte JS (2018) An efficient Bayesian meta-analysis approach for studying cross-phenotype genetic associations. PLoS Genet 14(2):e1007139
Mamidi TKK, Wu J, Hicks C (2019) Integrating germline and somatic variation information using genomic data for the discovery of biomarkers in prostate cancer. BMC Cancer 19(229):1–12
Mannino GC, Greco A, De Lorenzo C, Andreozzi F, Marini MA, Perticone F et al. (2013) A fasting insulin-raising allele at IGF1 locus is associated with circulating levels of IGF-1 and insulin sensitivity. PLoS ONE 8(12):e85483
Meng F, Yuan G, Zhu X, Zhou Y, Wang D, Guo Y (2018) Functional Variants Identified Efficiently through an Integrated Transcriptome and Epigenome Analysis. Sci Rep 8:2959.
Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J (2017) Differences between germline and somatic mutation rates in human and mice. Nat Commun 8:15183.
Mortezaei Z, Lanjanian H, Masoudi-nejad A (2017) Candidate novel long noncoding RNAs, MicroRNAs and putative drugs for Parkinson’s disease using a robust and efficient genome-wide association study. Genomics 109(3-4):158–164
Mortezaei Z, Cazier J-B, Mehrabi AA, Cheng C, Masoudi-Nejad A (2019) Novel putative drugs and key initiating genes for neurodegenerative disease determined using network ‐ based genetic integrative analysis. J Cell Biochem 120(4):5459–5471
Mortezaei Z, Tavallaei M (2021) Novel directions in data pre-processing and genome-wide association study (GWAS) methodologies to overcome ongoing challenges. Inform Med Unlocked 24(100586):2352–9148. ISSN
Mortlock S, Kendarsari RI, Fung JN, Gibson G, Yang F, Restuadi R et al. (2020) Tissue specific regulation of transcription in endometrium and association with disease. Hum Reprod 35(2):377–393
Nelson CS, Beck JN, Wilson KA, Plicher ER, Kapahi P, Brem RB (2016) Cross-phenotype association tests uncover genes mediating nutrient response in Drosophila. BMC Genom 17(1):867
Nguyen TV, Eisman JA (2020) Post-GWAS Polygenic Risk Score: Utility and Challenges. JBMR Plus. 4(11):e10411
Nica AC, Dermitzakis ET (2013) Expression quantitative trait loci: present and future. Philos Trans R Soc Lond B Biol Sci 368(1620):20120362
O’Reilly PF, Hoggart CJ, Pomyen Y, Calboli FCF, Elliott P, Jarvelin M-R et al. (2012) MultiPhen: joint model of multiple phenotypes can increase discovery in GWAS. PLoS ONE 7(5):e34861
Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC et al. (2015) Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet 47(4):381–386
Osgood JA, Knight JC (2018) Translating GWAS in rheumatic disease: approaches to establishing mechanism and function for genetic association with ankylosing spondylitis. Brief Funct Genom 17(5):308–318
Peat G, Jones W, Nuhn M, Marugán JC, Newell W, Dunham I et al. (2020) The open targets post-GWAS analysis pipeline. Bioinformatics. 36(9):2936–2937
Perrotti PP, Aterido A, Fernández-Nebro A, Cañete JD, Ferrándiz C, Tornero J et al. (2017) Genetic variation associated with cardiovascular risk in autoimmune diseases. PLoS ONE 12(10):e0185889
Petti AA, Williams SR, Miller CA, Fiddes IT, Srivatsan SN, Chen DY et al. (2019) A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat Commun 10:3660
Pierce SE, Booms A, Prahl J, van der Schans EJC, Tyson T, Coetzee GA (2020) Post-GWAS knowledge gap: the how, where, and when. npj Parkinsons Dis 6(23):2020
Plun-Favreau H, Lewis PA, Hardy J, Martins M, Wood NW (2010) Cancer and Neurodegeneration: between the Devil and the Deep Blue Sea. PLoS Genet 6(12):e1001257
Polster R, Petropoulos CJ, Bonhoeffer S, Guillaume F (2016) Epistasis and Pleiotropy Affect the Modularity of the Genotype-Phenotype Map of Cross-Resistance in HIV-1. Mol Biol Evol 33(12):3213–3225
Porcu E, Rueger S, Lepik K, eQTLGen Consortium, BIOS ConsortiumSantoni FA et al. (2019) Mendelian randomization in integrating GWAS and eQTL data reveals genetic determinants of complex and clinical traits. Nature Commun 10:3300.
Przybyla P, Shardlow M, Aubin S, Bossy R, de Castilho RE, Piperidis S et al. (2016) Text mining resources for the life sciences. Database (Oxford) 2016:baw145
R Core Team (2012) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org/.
Rai SN, Wu X, Srivastava DK, Craycroft JA, Rai JP, Srivastava S et al. (2018) Review: propensity score methods with application to the HELP clinic clinical study. Open Access Med Statistics 8:11–23
Ray D, Chatterjee N (2020) Effect of non-normality and low count variants on cross-phenotype association tests in GWAS. Eur J Hum Genet 28:300–312
Ren X, Kang B, Zhang Z (2018) Understanding tumor ecosystems by single-cell sequencing: promises and limitations. Genome Biol 19(1):211
Reimand J, Bader GD (2013) Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol Syst Biol 9(1):637
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–423
Rivandi M, Martens JWM, Hollestelle A (2018) Elucidating the Underlying Functional Mechanisms of Breast Cancer Susceptibility Through Post-GWAS Analyses. Front Genet 9:280
Romanoski CE, Glass CK, Stunnenberg HG, Wilson L, Almouzni G (2015) Epigenomics: roadmap for regulation. Nature. 518(7539):314–316
Saccenti E, Hoefsloot HCJ, Smilde AK, Westerhuis JA, Hendriks M, MWB (2014) Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics 10:361–374
Salinas YD, Wang Z, DeWan AT (2017) Statistical Analysis of Multiple Phenotypes in Genetic Epidemiologic Studies: From Cross-Phenotype Associations to Pleiotropy. Am J Epidemiol 187(4):855–863
Salomon MP, Li WLS, Edlund CK, Morrison J, Fortini BK, Win AK et al. (2016) GWASeq: targeted re-sequencing follow up to GWAS. BMC Genom 17:176
Schaid DJ, Chen W, Larson NB (2018) From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet 19(8):491–504
Schifano ED, Li L, Christiani DC, Lin X (2013) Genome-wide association analysis for multiple continuous secondary phenotypes. Am J Hum Genet 92(5):744–759
Shim H, Chasman DI, Smith JD, Mora S, Ridker PM, Nickerson DA et al. (2015) A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS ONE 10(4):e0120758
Slim L, Chatelain C, Azencott C-A, Vert J-P (2020) Novel Methods for Epistasis Detection in Genome-Wide Association Studies. PLoS ONE 15(11):e0242927
Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI et al. (2016) Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533(7601):95–99
Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW (2013) Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet 14(7):483–495
Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D (2019) Benefits and limitations of genome-wide association studies. Nat Rev Genet 20:467–484
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N et al. (2018) COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47(D1):D941–D947
Tettey P, Simpson Jr S, Taylor BV, van der Mei IAF (2015) The co-occurrence of multiple sclerosis and type 1 diabetes: shared aetiologic features and clinical implication for MS aetiology. J Neurol Sci 348(1-2):126–131
The Brainstorm Consortium (2018) Analysis of Shared Heritability in Common Disorders of the Brain. Science 360:6395
The Gene Ontology Consortium (2019) The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 47(D1):D330–D338
The GTEx Consortium (2013) The Genotype-Tissue Expression (GTEx) project. Nat Genet 45(6):580–585
The UniProt Consortium (2018) UniProt: the universal protein knowledgebase. Nucleic Acids Res 46:2699
Thompson PM, Jahanshad N, Ching CRK, Salminen LE, Thomopoulos SI, Bright J et al. (2020) ENIGMA and global neuroscience: a decade of large-scale studies of the brain in health and disease across more than 40 countries. Transl Psychiatry 10(1):100
Turnbull C, Ahmed S, Morrison J, Pemet D, Renwick A, Maranian M et al. (2010) Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet 42(6):504–507
Ung MH, Liu C-C, Cheng C (2016) Integrative analysis of cancer genes in a functional interactome. Sci Rep 6(1):29228
Verma A, Lucas A, Verma SS, Zhang Y, Josyula N, Khan A et al. (2018) PheWAS and Beyond: the Landscape of Associations with Medical Diagnoses and Clinical Measures across 38,662 Individuals from Geisinger. Am J Hum Genet 102(4):592–608
Verma A, Bang L, Miller JE, Zhang Y, Lee MTM, Zhang Y et al. (2019) Human-Disease Phenotype Map Derived from PheWAS across 38,682 Individuals. The Am J of Hum Genet 104(1):55–64
Wang M, Roux F, Batoli C, Huard-Chauveau C, Meyer C, Lee H et al. (2018) Two-way mixed-effects methods for joint association analysis using both host and pathogen genomes. Proc Natl Acad Sci 115(24):E5440–E5449
Wang Q-L, Tan W-L, Zhao Y-L, Shao M-M, Chu J-H, Huang X-D et al. (2016) Data analysis in the post-genome-wide association study era. Chronic Dis Transl Med 2(4):231–234
Wang QS, Kelley DR, Ulirsch J, Kanai M, Sadhuka S, Cui R et al. (2021) Leveraging supervised learning for functionally informed fine-mapping of cis-eQTLs identifies an additional 20,913 putative causal eQTLs. Nat Commun 12(1):3394
Wang W, Xu ZZ, Costanzo M, Boone C, Lang CA, Myers CL (2017) Pathway-based discovery of genetic interactions in breast cancer. PLoS Genet 13(9):e1006973
Ward LD, Kellis M (2011) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 40(D1):D930–D934
Watanabe K, Taskesen E, van Bochoven A, Posthuma D (2017) Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8(1):1826
Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Cancer Genome Atlas Research Network, et al. (2013) The cancer genome atlas pan-cancer analysis project Nat Genet 45(10):1113–1120
Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447(7145):661–678
Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H et al. (2014) The NHGRI GWAS catalog: a curated resource of snp-trait associations. Nucleic Acids Res 42(D1):D1001–D1006
Weng M-P, Liao B-Y (2010) MamPhEA: a web tool for mammalian phenotype enrichment analysis. Bioinformatics 26(17):2212–2213
Wu GD, Compher C, Chen EZ, Smith SA, Shah RD, Bittinger K et al. (2016) Comparative metabolomics in vegans and omnivores reveal constraints on diet-dependent gut microbiota metabolite production. Gut 65(1):63–72
Wu J, Mamidi TKK, Zhang L, Hicks C (2019) Integrating Germline and Somatic Mutation Information for the Discovery of Biomarkers in Triple-Negative Breast Cancer. Int J Environ Res Public Health 16(6):1055
wwPDB Consortium (2019) Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res 47:D520–D528
Xiang R, van den Berg I, Macleod IM, Daetwyler HD, Goddard ME (2020) Effect direction meta-analysis of GWAS identifies extreme, prevalent and shared pleiotropy in a large mammal. Commun Biol 3:88
Yang X, Zhang S, Sha Q (2019) Joint Analysis of Multiple Phenotypes in Association Studies based on Cross-Validation Prediction Error. Sci Rep 9:1073
Zhang X, Veturi Y, Verma S, Bone W, Verma A, Lucas A et al. (2019) Detecting potential pleiotropy across cardiovascular and neurological diseases using univariate, bivariate, and multivariate methods on 43,870 individuals from the eMERGE network. Pac Symp Biocomput 24:272–283
Zhao S, Jing W, Samuels DC, Sheng Q, Shyr Y, Guo Y (2017) Strategies for processing and quality control of Illumina genotyping arrays. Brief Bioinform 19(5):765–775
Zhao H, Mitra N, Kanetsky PA, Nathanson KL, Rebbeck TR (2018) A practical approach to adjusting for population stratification in genome-wide association studies: principal components and propensity scores (PCAPS). Stat Appl Genet Mol Biol 17(6):1–20
Zhao J, Sauvage C, Zhao J, Bitton F, Bauchet G, Liu D et al. (2019a) Meta-analysis of genome-wide association studies provides insights into genetic control of tomato flavor. Nat Commun 10(1534):1–12
Zhao T, Hu Y, Zang T, Wang Y (2019b) Integrate GWAS, eQTL, and mQTL Data to Identify Alzheimer’s Disease-Related Genes. Front Genet 10:1021
Zhou Z, Stephens M (2014) Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat Methods 11(4):407–409
Zhou X, Li D, Zhang B, Lowdon RF, Rockweiler NB, Sears RL et al. (2015) Epigenomic annotation of genetic variants using the Roadmap Epigenome Browser. Nat Biotechnol 33(4):345–346
Acknowledgements
We thank Dr Frank Hailer for careful and thorough reading of our paper, thoughtful and insightful comments and constructive suggestions, which helped to improve the quality of our paper. We also thank anonymous reviewers for their suggestions and comments that were useful for improving our paper’s presentation.
Author information
Authors and Affiliations
Contributions
ZM and MT came up with the concept of the review. ZM reviewed and interpreted the literature and drafted the paper. ZM and MT critically revised the paper. ZM approved the final version of the review.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mortezaei, Z., Tavallaei, M. Recent innovations and in-depth aspects of post-genome wide association study (Post-GWAS) to understand the genetic basis of complex phenotypes. Heredity 127, 485–497 (2021). https://doi.org/10.1038/s41437-021-00479-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-021-00479-w
