
Abstract
Vascular aging is a major contributing factor to cardiovascular disease. The aged blood vessels, characterized by vascular wall thickening and stiffening, are instigated by endothelial cell dysfunction induced by oxidative stress and inflammation. von Willebrand Factor (vWF) is a glycoprotein known for its role in coagulation, and plasma levels of vWF are increased with age. Elevated vWF promotes thrombosis, atherosclerotic plaque formation, inflammation and proliferation of vascular smooth muscle cells. Cadmium (Cd) is an environmental pollutant associated with increased morbidity and mortality of cardiovascular disease. At low concentrations, Cd activates pro-survival signaling in endothelial cells, however enhances intima-media thickness and atherogenesis. A non-cytotoxic dose of Cd also increases endothelial vWF expression and secretion in vivo and in vitro. In this review, we summarize the molecular mechanisms underlying vWF-promoted vascular aging-associated pathologies and Cd-induced vWF expression. In addition, we propose that exposure to low-dose Cd is a risk factor for vascular aging, through elevation of plasma vWF.
Similar content being viewed by others

Vascular aging
Vascular aging, the decline in vascular structure and function with age, contributes to cardiac and peripheral vascular diseases1. In aging vessels, increased intima-media thickness (IMT) occurs linearly (~5 μm year–1) with older age, accompanied by vascular smooth muscle cell (VSMC) proliferation, and resultant increase of subendothelial extracellular matrix (ECM)2,3. The aged vessels also exhibit elastin fragmentation and calcification, leading to increased vessel stiffness and reduced compliance4. Age-related mechanical and adhesive properties of vascular cells also increases vascular stiffness, which destabilizes endothelium and subsequently promotes leukocyte transmigration and atherosclerosis5,6.
Vascular endothelial cell (EC) dysfunction is an early manifestation of vascular aging that precedes the structural remodeling and stiffness4. In the elderly, EC injury leads to malfunction of vascular tone, vascular permeability, and hemostasis. Nitric oxide (NO) is a major vasodilator released by ECs, and decrease of NO production impairs endothelium-dependent dilation (EDD) in vascular aging7. Age-dependent blood-brain barrier breakdown occurs initially in the hippocampus, a region critical for learning and memory8. In addition, endothelium-derived anticoagulant proteins including prostacyclin and thrombomodulin are down-regulated in vascular aging9. Whereas the expression of procoagulants such as von Willebrand Factor (vWF), thromboxane A2 and plasminogen activator inhibitor-1 are increased, favoring the development of thrombosis in vascular aging10,11,12.
Oxidative stress and inflammation are strongly associated with age-related endothelial dysfunction and vascular stiffening13. Aging is accompanied with increased production of reactive oxygen species (ROS) by reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidases and mitochondria, which reduces NO bioavailability and impairs EDD14. Oxidative stress also activates proinflammatory signaling pathways, including nuclear factor-κB (NF-κB), and increases proinflammatory cytokine gene expression including tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1) and interleukin-615,16. Both inflammatory cytokines and ROS induce the secretion of matrix metalloproteinase and transforming growth factor β1 that mediate ECM remodeling including elastin fragmentation, collagen accumulation and calcification17. Increased NF-κB activity upregulates the expression of endothelial adhesion molecules to facilitate leukocyte adhesion and transmigration15. Immune cells infiltration exacerbates the presence of inflammatory cytokines and the production of ROS, thus creating a vicious feed-forward cycle to accelerate vascular aging13. In addition, genomic instability, epigenetic alterations, deregulated nutrient sensing, stem cell dysfunction, and autophagy may involve in the development of vascular aging, and have been summarized in recent reviews18,19.
Cadmium (Cd) is a well-known hazardous pollutant20. The vascular endothelium is one of the major targets of Cd21. Epidemiological studies suggest adverse effects of environmental Cd exposure on age-related cardiovascular diseases in the general population, raising concerns about the validity of the current safe intake level22,23,24,25. Our group and others demonstrated that low-dose Cd upregulates endothelial expression of vWF, a key contributor of vascular aging-associated pathologies26,27. Therefore, we propose that environmental Cd exposure is a risk factor for vascular aging, possibly by elevation of vWF expression.
vWF
vWF is a multimeric glycoprotein expressed in ECs and megakaryocytes28. ECs are the primary source of plasma and subendothelial vWF. Following synthesis in ECs, vWF is either constitutively secreted or stored in Weibel-Palade bodies (WPBs), from where it is released into plasma and basement membrane upon activation28. Increased plasma vWF is a hallmark of EC dysfunction. Plasma vWF regulates hemostasis by facilitating platelet adhesion and coagulation factor VIII (FVIII) stabilization29,30. Subendothelial vWF mediates initial attachment of platelet to the basement membrane after EC injury31. Deficiency of vWF, either quantitative or qualitative, causes a bleeding disorder known as von Willebrand disease (VWD)32.
Plasma vWF level and vWF expression in ECs is increased with age. Plasma vWF antigen shows a gradual increase of 1-2% per year in healthy human, and is accompanied by enhanced functional activity10,33,34. A substantial increase in vWF levels is found in both men and women after midlife (above 40 years of age)33. Increased plasma vWF levels have also been observed in patients with type 1 VWD where vWF level is normalized with advancing age, but not in type 2 and 3 VWD35. Notably, immunohistochemical analysis of cellular vWF expression demonstrated significantly elevated staining intensity of vWF in lung vasculature in adults compared to that in children36. In addition, age-related increase in vWF expression in brain ECs and in hepatic sinusoidal ECs have been documented in mice, rats, human, and non-human primates37,38,39.
The roles of vWF in vascular aging-associated vascular pathologies
vWF is involved in several age-related vascular pathologies. Plasma levels of vWF are positively correlated with IMT and arterial stiffness in human cohorts40,41. In addition, higher vWF levels are associated with artery calcification and ischemic stroke42. Antibodies against vWF improve EC functions in animal models of vascular injury and patients with stable angina43,44.
vWF alters its conformation in response to hydrodynamic forces in the bloodstream45,46. The shear stress is proportional to blood flow velocity and viscosity, and inversely proportional to the blood vessel diameter47. Variations in shear stress and hydrodynamic forces occur near arterial bifurcations, branch mouths, and curvatures, which are associated with age-associated vascular remodeling47. Under elevated shear stress flow conditions, the structure of vWF is altered from a globular form (collapsed conformation) to a stretched linear conformation46. This structural transition correlates with the increased vWF adhesiveness to collagen and platelets45. A pathological increase in the adhesiveness of vWF to platelets in the blood circulation leads to thrombosis, thrombotic thrombocytopenia, and organ failure45,48.
vWF promotes thrombosis
Vascular aging increases the risk to develop venous and arterial thrombosis in the older population1,49. Age-associated production of vWF may cause increased thrombogenicity50. After detachment of aged ECs from the vascular wall, subendothelial vWF is exposed to the bloodstream, and directly initiates platelet adhesion to the subendothelial tissue by interaction with platelet glycoprotein (GP) Ib31. Plasma vWF also binds to the subendothelial collagen and facilitates platelet adhesion51. The vWF-mediated low-affinity adhesion induces the activation of platelet αIIbβ3 integrin that in turn binds to fibrinogen, fibrin and vWF to stabilize platelet adhesion29. vWF immobilized on adherent activated platelets provides attachment sites for additional circulating platelets, facilitating platelet aggregation and thrombus formation29. Under high shear rates, platelet adhesion and aggregation are more dependent on vWF52. At shear rates over 1,0000/s, a condition that occurs only in stenotic arteries, thrombus formation is exclusively mediated by vWF-GPIb interaction52. In addition, plasma FVIII levels increase with age, and this increase is dependent on vWF levels34. In circulation, FVIII forms a complex with vWF, which protects FVIII from proteolytic degradation30. Upon vascular injury, activated FVIII (FVIIIa) dissociates from vWF, and binds to FIXa on the phospholipid membrane of activated platelets to form the factor Xase complex53. FVIIIa within the factor Xase complex stabilizes the FIXa active site, and serves as a molecular bridge between FIXa and its substrate FX, facilitating FXa generation53. FXa in complex with FVa activates prothrombin to thrombin54. Thrombin cleaves fibrinogen and yields monomeric fibrin, which deposits and polymerizes to stabilize the platelet-rich thrombus54. vWF also binds to the apical surface of activated ECs, where it mediates platelet adhesion on intact endothelial surface and facilitates thrombus formation in the absence of EC injury55. Thus, increased vWF in the microenvironment of aged ECs may promote platelets aggregation and subsequent thrombus formation.
vWF modulates vascular remodeling
In aged blood vessels, VSMCs switch from a quiescent contractile phenotype to a synthetic phenotype characterized by increased cell proliferation, migration, and production of ECM proteins, promoting intimal thickening3. vWF is constitutively released into subendothelial space28, and plasma vWF penetrates into the intima of vessel walls through intercellular gaps caused by ECs injury56,57. Age-related accumulation of vWF is observed in porcine aortic valve subendothelium, and associated with valvular interstitial cell calcification58. In ligation-induced carotid intimal hyperplasia, vWF expression in ECs and vWF deposition in neointimal ECM is significantly elevated56. The level of vWF is positively correlated with the degree of intimal hyperplasia56. Increased expression and deposition of vWF in hyperplastic intima is also noted in atherosclerotic plaques, vascular grafts, and balloon angioplasty57,59,60,61. vWF deficiency in mice leads to a decreased outward remodeling and VSMC proliferation62. In vitro studies show that vWF directly increases VSMC proliferation and migration with a dose-response effect56,63. vWF interacts with integrin αvβ3 on VSMC to facilitate VSMC adhesion to the endothelial basement membrane64. In addition, vWF binds to the LRP4-receptor on VSMC, which in turn triggers integrin αvβ3 signaling to promote VSMC proliferation63. Blockage of αvβ3 signaling inhibits the adhesion and proliferation of VSMC induced by vWF63,64.
vWF facilitates vascular inflammation
Chronic, low-grade inflammation in the vascular wall is a hallmark of vascular aging13. As a critical early step in chronic vascular inflammation, leukocyte-endothelial interaction is increased in aging vessels due to enhanced endothelial adhesiveness65. The leukocyte subsequently releases cytokines, proteases and ROS, which induce vascular aging39,65. vWF promotes leukocyte-endothelial interaction via multiple mechanisms. vWF directs biogenesis of WPBs that contain P-selectin, a known inflammation mediator66. vWF deficiency impairs P-selectin translocation to EC surface and reduces leukocyte recruitment in early phases of inflammation67. In addition, endothelium-associated vWF creates an adhesive surface for neutrophils and mediates their rolling through P-selectin glycoprotein ligand (PSGL)-1 and stable adhesion through β2-integrins under static and low-shear conditions68. With a high shear stress in arteries and arterioles, endothelium-associated vWF captures platelets which promote neutrophil rolling via P-selectin/PSGL-1 and neutrophil adhesion via GPIb/Macrophage (MAC)-1 interactions69. Moreover, vWF promotes neutrophil extravasation in a strictly platelet and GPIb dependent way70. Subendothelial vWF inhibits tight junction protein claudin-5 expression thus destabilizes endothelial barrier71. vWF also promotes alternative complement pathway activation and stabilizes neutrophil extracellular traps (NETs), both conditions effect age-related vascular inflammation72,73,74. Notably, anti-vWF treatment showed a vascular anti-inflammatory effect both in prophylactic and therapeutic administration75.
vWF enhances atherosclerosis
Aging is an independent risk factor for the development of atherosclerosis1, and age-associated vascular wall degeneration is also aggravated by the presence of atherosclerosis76. In population-based stuides, plasma levels of vWF are markedly higher in patients with atherosclerosis, and vWF are positively correlated with the plaque thickness and stenosis area77,78. In carotid and coronary arteries from human, large numbers of WPBs are present in ECs at sites of atherosclerotic lesions79. Increased vWF staining is also observed in the intima of human atherosclerotic arteries63,80. Although clinical reports are inconsistent on whether VWD patients are protected from atherosclerosis81,82,83,84, pigs with severe VWD show a decreased number of aortic plaques on a cholesterol-rich diet and mice deficient of vWF display reduced fatty streaks85,86. Endothelial vWF is up-regulated in response to hypercholesterolemia before the advent of fatty streaks and recruits circulating platelets to the endothelium at atherosclerosis-prone sites87. vWF-bound platelets interact with monocytic PSGL-1 via P-selectin and with monocytic MAC-1 via GPIb, thereby promoting monocytes attachment on endothelium and transmigration69. Recruited monocytes in subendothelium differentiate into macrophages to engulf oxidized low-density lipoprotein (oxLDL) by scavenger receptor, which gradually leads to accumulation of LDL-derived cholesterol and subsequent lipid-rich core formation76. vWF deficiency reduces the presence of monocytes in the fatty streaks86. Hence, vWF may facilitate monocytes transmigration to support the development of atherosclerotic lesions.
vWF is a multitasker in cellular processes, and the resulting pathophysiological consequences of vWF dysregulation are still controversial. Further investigation is needed to clarify the detailed roles and the underlying mechanisms of vWF in vascular aging-associated pathologies.
Low-dose Cd induces endothelial vWF expression and secretion
A positive correlation between cigarette smoking, a primary source of Cd exposure, and plasma vWF levels has been detected88. Low-dose Cd (<10 μM) does not induce ECs death, but increases VEGF receptor-2 expression and promotes angiogenesis89. The effect of low-dose Cd exposure on endothelial vWF expression and secretion was examined26. Experimental mice were fed with cadmium chloride (CdCl2) in distilled water and displayed elevated expression of vWF in the endothelium of lung and kidney. In vitro, CdCl2 at concentrations as low as 1 μM (the rough concentration found in the arterial intima of smokers were 1.5 μM90) induces the expression and secretion of vWF in human umbilical vein endothelial cells (HUVECs). Mechanistically, CdCl2 exposure increases ETS-related gene (ERG) expression and enhances its binding to the -56 ETS-motif on the promoter of the vWF gene (vWF), and thereby elevates vWF transcription26. ERG, a member of the ETS family of transcription factors, is specifically expressed in ECs91. The cis-acting element GGAA/T, situated at -56 in the core promoter of vWF, is bound and activated by ERG92. Interference of ERG expression reduces vWF expression and this effect is abolished by mutations of the -56 ETS-motif of the vWF promoter92. Although ERG is known for its role in endothelial homeostasis, aberrant expression of endothelial ERG has been observed in pathological conditions including vascular malignancies93,94,95,96,97, arterial calcification98, and tumour neovascularization99,100. In addition, overexpression of ERG in the Xenopus embryo results in developmental defects and ectopic endothelial differentiation101.
The human vWF promoter contains multiple cis-regulatory sequences that positively or negatively regulate gene expression. A GATA transcription factor binding site, situated at position +220 in the first exon of vWF, mediates vWF transcription in an ECs-specific manner102. Transcription factor NF-κB interacts with the -1793 sequence to repress vWF expression103. In contrast to ERG, the levels of GATA3 and NF-κB are not effected by low-dose Cd26. Nevertheless, other mechanisms may also mediate low-dose Cd induced vWF expression, including nuclear factor 1-like protein, histone H1-like protein and nuclear factor-Y transcription factor, which bind to the core promoter of vWF104,105,106.
Environmental Cd exposure is a risk factor for vascular aging
Cd is a toxic metal that occurs naturally in sulfide ores107. Since the 1940s, Cd has been widely utilized in production of batteries, alloys, coatings, plating, and plastic stabilizers20. In addition, large amount of Cd is released into environment through vehicle exhaust, pesticides and fertilizers20. Currently, Cd is ranked the seventh in environmental toxic pollutants108. For population without occupational exposure, environmental Cd enters human body primarily through ingestion of Cd-contaminated food and water20. Environmental Cd exposure is particularly high in East Asian due to the dietary exposure via consumption of Cd-contaminated rice, fish, and shellfish109. Cigarette smoking is the dominant source of inhaled Cd in addition to ambient air pollution20. After absorption, Cd is distributed throughout the body via bloodstream, progressively accumulating mainly in the kidney and liver110.
Cd is transported in blood plasma bound to proteins or as free ions21. The U.S. OSHA safety standard is currently 5 μg/L (44.5 nmol/L) for blood Cd (bCd)111. In the general US population, bCd at even low levels are associated with increased prevalence of peripheral artery disease (PAD)23. Tellez-Plaza et al. reported that the prevalence of PAD increases with bCd (within 1 μg/L) in a dose-dependent manner25. In a 5-year follow-up of 64-year-old women from Sweden, prevalence of PAD is significantly higher in the group with high bCd (0.44 to 4.07 μg/L) than that with low bCd (0.08 to 0.25 μg/L)24. In addition, low-level Cd exposure of general populations is associated with myocardial infarction, stroke, heart failure, and cardiovascular mortality112,113,114. In healthy young female subjects with an average age of 20.6 years, plasma levels of Cd are positively correlated to IMT115. Environmental Cd exposure is also associated with atherosclerotic plaques development in middle-aged men and women from Sweden22. Cd-induced atherosclerotic changes have been reported in the coronary arteries of rabbits, and the aorta of Wistar rats and White Carneau pigeons116,117,118,119. In addition, Cd-fed ApoE−/− mice exhibit significantly increased area of aortic plaques115,120,121. These evidences suggest that, Cd exposure, even at relatively low levels, may increase vascular aging-associated pathologies.
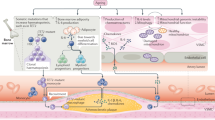
The vascular endothelium lines the lumenal surface of all blood vessels and form the capillary networks that deliver oxygen and nutrients to tissues of the body4. As a toxicant circulating in blood, Cd interacts directly with vascular ECs, thus targeting the vascular system21. Acute exposure to high-level Cd causes ECs apoptosis or necrosis and results in hemorrhage in various tissues21. Whereas at non-cytotoxic concentrations, Cd activates pro-survival signaling in ECs, leading to enhanced cell proliferation89. Low-dose Cd exposure does not induce toxicity of ECs, but significantly increases endothelial vWF expression and secretion26. In addition, Cd treatment increases the number and exocytosis of WPBs, the storage receptacle of vWF, in ECs of thoracic aorta of rats27. Therefore, environmental Cd exposure increases vWF expression, and may subsequently accelerate vascular aging (Fig. 1). However, the role of Cd exposure on vWF-dependent vascular pathologies, e.g., platelet adhesion, shall be validated in the future studies.

Low-dose Cd exposure induces the expression of ETS-related gene (ERG), which transcriptionally active von Willebrand Factor (vWF) expression and secretion. At sites of endothelial injury, plasma and subendothelial vWF interacts with circulating platelets via GP Ib and integrin αIIbβ3 to promote platelet adhesion and aggregation. Plasma vWF also binds to intact but activated endothelium, where it facilitates platelets binding under flow and intravascular thombus formation. vWF bound platelets are activated to assist the rolling and attachment of monocytes to the endothelium via P-selectin, and to promote monocyte extravasation via GPIb binding. Recruited monocytes differentiate into macrophages to engulf oxidized low-density lipoprotein (oxLDL), leading to the accumulation of LDL-derived cholesterol and lipid-rich core formation. vWF also mediates neutrophil attachment and emigration through platelet activation or direct interaction with neutrophils via P-selectin glycoprotein ligand (PSGL)-1 and Macrophage (MAC)-1, resulting in a proinflammatory state of the endothelium. In addition, subendothelial vWF promotes vascular smooth muscle cells (VSMCs) proliferation and migration in an αvβ3-dependent manner, leading to increased intima-media thickness and stiffness.
Discussion
Vascular aging is associated with inflammation and oxidative stress, both of which increase vWF secretion13. Although proinflammatory cytokines such as TNF-α and IL-1 induces oxidative stress and stimulates vWF secretion by the stimulation of exocytosis of WPBs, they potently inhibit the expression of vWF, leading to a transient elevation but long-term down-regulation of plasma vWF122,123. NF-κB signaling mediates oxidative stress and is activated by endogenous hydrogen peroxide (H2O2) during aging15, but activation of NF-κB signaling also represses vWF expression103. Although a high concentration of exogenous H2O2 (≥ 200 μM) increases endothelial vWF expression, it induces substantial cell death in ECs and is unlikely to be reached in human pathophysiology124. Cd at a concentration lower than 10 μM does not induce cell death but activates pro-survival signaling and increases proliferation of ECs89. Low-dose Cd-induced vWF transcription may constantly produce additional vWF protein to sustain age-associated increase of vWF levels.
Plasma vWF levels are increased with aging. Elevated vWF induces vascular aging-associated pathologies by promotion of thrombus formation, vascular remodeling, vascular inflammation and atherogenesis. Cd is a widespread environmental pollutant, and associated with age-related vascular diseases. At non-cytotoxic concentrations, Cd induces the expression and secretion of vWF in ECs. Therefore, environmental Cd exposure may accelerate vascular aging through the elevation of endothelial vWF. Reduction of Cd exposure and therapeutic approaches to decrease vWF expression may decelerate vascular aging.
Data availability
All data here disclosed are published in the literature as indicated in the references section.
References
Lakatta, E. G. & Levy, D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation 107, 139–146 (2003).
Homma, S., Hirose, N., Ishida, H., Ishii, T. & Araki, G. Carotid plaque and intima-media thickness assessed by b-mode ultrasonography in subjects ranging from young adults to centenarians. Stroke 32, 830–835 (2001).
Lacolley, P., Regnault, V., Nicoletti, A., Li, Z. & Michel, J. B. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc. Res. 95, 194–204 (2012).
Tesauro, M. et al. Arterial ageing: from endothelial dysfunction to vascular calcification. J. Intern. Med. 281, 471–482 (2017).
Starodubtseva, M. N. Mechanical properties of cells and ageing. Ageing Res. Rev. 10, 16–25 (2011).
Huynh, J. et al. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci. Transl. Med. 3, 112ra122 (2011).
Lyons, D., Roy, S., Patel, M., Benjamin, N. & Swift, C. G. Impaired nitric oxide-mediated vasodilatation and total body nitric oxide production in healthy old age. Clin. Sci. (Lond) 93, 519–525 (1997).
Montagne, A. et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302 (2015).
Komatsumoto, S., Onoda, A. & Nara, M. [Changes in the level of thrombomodulin with aging]. Nihon Ronen Igakkai Zasshi 33, 512–517 (1996).
Kokame, K., Sakata, T., Kokubo, Y. & Miyata, T. von Willebrand factor-to-ADAMTS13 ratio increases with age in a Japanese population. J. Thromb. Haemost. 9, 1426–1428 (2011).
Tang, E. H. & Vanhoutte, P. M. Gene expression changes of prostanoid synthases in endothelial cells and prostanoid receptors in vascular smooth muscle cells caused by aging and hypertension. Physiol. Genomics 32, 409–418 (2008).
Hashimoto, Y. et al. Relationship between age and plasma t-PA, PA-inhibitor, and PA activity. Thromb. Res. 46, 625–633 (1987).
El Assar, M., Angulo, J. & Rodríguez-Mañas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 65, 380–401 (2013).
Csiszar, A. et al. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 90, 1159–1166 (2002).
Ungvari, Z. et al. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am. J. Physiol. Heart Circ. Physiol. 293, H37–H47 (2007).
Donato, A. J. et al. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ. Res. 100, 1659–1666 (2007).
Wang, M. et al. Chronic matrix metalloproteinase inhibition retards age-associated arterial proinflammation and increase in blood pressure. Hypertension 60, 459–466 (2012).
Ungvari, Z., Tarantini, S., Sorond, F., Merkely, B. & Csiszar, A. Mechanisms of vascular aging, a geroscience perspective: JACC focus seminar. J. Am. Coll. Cardiol. 75, 931–941 (2020).
Donato, A. J., Machin, D. R. & Lesniewski, L. A. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ. Res. 123, 825–848 (2018).
Zhang, H. & Reynolds, M. Cadmium exposure in living organisms: a short review. Sci. Total Environ. 678, 761–767 (2019).
Prozialeck, W. C., Edwards, J. R. & Woods, J. M. The vascular endothelium as a target of cadmium toxicity. Life Sci. 79, 1493–1506 (2006).
Fagerberg, B. et al. Cadmium exposure and atherosclerotic carotid plaques–results from the Malmö diet and Cancer study. Environ. Res. 136, 67–74 (2015).
Navas-Acien, A. et al. Lead, cadmium, smoking, and increased risk of peripheral arterial disease. Circulation 109, 3196–3201 (2004).
Fagerberg, B., Bergström, G., Borén, J. & Barregard, L. Cadmium exposure, intercellular adhesion molecule-1 and peripheral artery disease: a cohort and an experimental study. BMJ Open 3, e002489 (2013).
Tellez-Plaza, M., Navas-Acien, A., Crainiceanu, C. M., Sharrett, A. R. & Guallar, E. Cadmium and peripheral arterial disease: gender differences in the 1999-2004 US National Health and Nutrition Examination Survey. Am. J. Epidemiol. 172, 671–681 (2010).
Wang, X. et al. Low dose cadmium upregulates the expression of von Willebrand factor in endothelial cells. Toxicol. Lett. 290, 46–54 (2018).
Doi, Y. et al. Increase in number of Weibel-Palade bodies and endothelin-1 release from endothelial cells in the cadmium-treated rat thoracic aorta. Virchows Arch. 428, 367–373 (1996).
Lenting, P. J., Christophe, O. D. & Denis, C. V. von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood 125, 2019–2028 (2015).
Ruggeri, Z. M. & Mendolicchio, G. L. Adhesion mechanisms in platelet function. Circ. Res. 100, 1673–1685 (2007).
Koppelman, S. J. et al. Requirements of von Willebrand factor to protect factor VIII from inactivation by activated protein C. Blood 87, 2292–2300 (1996).
Savage, B., Saldívar, E. & Ruggeri, Z. M. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 84, 289–297 (1996).
Favaloro, E. J. & Pasalic, L. Laboratory diagnosis of von Willebrand disease (VWD): geographical perspectives. Semin. Thromb. Hemost. 48, 750–766 (2022).
Davies, J. A., Hathaway, L. S., Collins, P. W. & Bowen, D. J. von Willebrand factor: demographics of plasma protein level in a large blood donor cohort from South Wales in the United Kingdom. Haemophilia 18, e79–e81 (2012).
Albánez, S. et al. Aging and ABO blood type influence von Willebrand factor and factor VIII levels through interrelated mechanisms. J. Thromb. Haemost. 14, 953–963 (2016).
Seaman, C. D. & Ragni, M. V. The effect of age on von Willebrand factor and bleeding symptoms in von Willebrand disease. Thromb. Haemost. 120, 1159–1165 (2020).
Müller, A. M., Skrzynski, C., Nesslinger, M., Skipka, G. & Müller, K. M. Correlation of age with in vivo expression of endothelial markers. Exp. Gerontol. 37, 713–719 (2002).
Hilmer, S. N. et al. Age-related changes in the hepatic sinusoidal endothelium impede lipoprotein transfer in the rat. Hepatology 42, 1349–1354 (2005).
McLean, A. J. et al. Age-related pseudocapillarization of the human liver. J. Pathol. 200, 112–117 (2003).
Yousef, H. et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat. Med. 25, 988–1000 (2019).
Karasek, D. et al. Prothrombotic markers in asymptomatic dyslipidemic subjects. J. Thromb. Thrombol. 31, 27–36 (2011).
Páramo, J. A., Beloqui, O., Colina, I., Diez, J. & Orbe, J. Independent association of von Willebrand factor with surrogate markers of atherosclerosis in middle-aged asymptomatic subjects. J. Thromb. Haemost. 3, 662–664 (2005).
Sonneveld, M. A. et al. Relationship of Von Willebrand Factor with carotid artery and aortic arch calcification in ischemic stroke patients. Atherosclerosis 230, 210–215 (2013).
Muller, O. et al. von Willebrand factor inhibition improves endothelial function in patients with stable angina. J. Cardiovasc. Transl. Res. 6, 364–370 (2013).
Gandhi, C., Motto, D. G., Jensen, M., Lentz, S. R. & Chauhan, A. K. ADAMTS13 deficiency exacerbates VWF-dependent acute myocardial ischemia/reperfusion injury in mice. Blood 120, 5224–5230 (2012).
Arce, N. A., Cao, W. & Brown, A. K. Activation of von Willebrand factor via mechanical unfolding of its discontinuous autoinhibitory module. Nat. Commun. 12, 2360 (2021).
Müller, J. P. et al. Force sensing by the vascular protein von Willebrand factor is tuned by a strong intermonomer interaction. Proc. Natl Acad. Sci. USA 113, 1208–1213 (2016).
Cunningham, K. S. & Gotlieb, A. I. The role of shear stress in the pathogenesis of atherosclerosis. Lab Invest. 85, 9–23 (2005).
Deng, W. et al. Platelet clearance via shear-induced unfolding of a membrane mechanoreceptor. Nat. Commun. 7, 12863 (2016).
Molnár, A. et al. The aging venous system: from varicosities to vascular cognitive impairment. Geroscience 43, 2761–2784 (2021).
Alavi, P. & Rathod, A. M. Age-associated increase in thrombogenicity and its correlation with von Willebrand factor. J. Clin. Med. 10, 4190 (2021).
Savage, B., Sixma, J. J. & Ruggeri, Z. M. Functional self-association of von Willebrand factor during platelet adhesion under flow. Proc. Natl Acad. Sci. USA 99, 425–430 (2002).
Jackson, S. P. The growing complexity of platelet aggregation. Blood 109, 5087–5095 (2007).
Saenko, E. L., Shima, M. & Sarafanov, A. G. Role of activation of the coagulation factor VIII in interaction with vWf, phospholipid, and functioning within the factor Xase complex. Trends Cardiovasc. Med. 9, 185–192 (1999).
Furie, B. & Furie, B. C. Molecular and cellular biology of blood coagulation. N. Engl. J. Med. 326, 800–806 (1992).
André, P. et al. Platelets adhere to and translocate on von Willebrand factor presented by endothelium in stimulated veins. Blood 96, 3322–3328 (2000).
Qin, F., Impeduglia, T., Schaffer, P. & Dardik, H. Overexpression of von Willebrand factor is an independent risk factor for pathogenesis of intimal hyperplasia: preliminary studies. J. Vasc. Surg. 37, 433–439 (2003).
Giddings, J. C., Banning, A. P., Ralis, H. & Lewis, M. J. Redistribution of von Willebrand factor in porcine carotid arteries after balloon angioplasty. Arterioscler. Thromb. Vasc. Biol. 17, 1872–1878 (1997).
Balaoing, L. R., Post, A. D., Liu, H., Minn, K. T. & Grande-Allen, K. J. Age-related changes in aortic valve hemostatic protein regulation. Arterioscler. Thromb. Vasc. Biol. 34, 72–80 (2014).
De Meyer, G. R. et al. Intimal deposition of functional von Willebrand factor in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 19, 2524–2534 (1999).
Qin, F. et al. Remodeling and suppression of intimal hyperplasia of vascular grafts with a distal arteriovenous fistula in a rat model. J. Vasc. Surg. 34, 701–706 (2001).
Bosmans, J. M. et al. Fibrin(ogen) and von Willebrand factor deposition are associated with intimal thickening after balloon angioplasty of the rabbit carotid artery. Arterioscler. Thromb. Vasc. Biol. 17, 634–645 (1997).
Laboyrie, S. L. & de Vries, M. R. von Willebrand factor: a central regulator of arteriovenous fistula maturation through smooth muscle cell proliferation and outward remodeling. J. Am. Heart Assoc. 11, e024581 (2022).
Lagrange, J. & Worou, M. E. The VWF/LRP4/αVβ3-axis represents a novel pathway regulating proliferation of human vascular smooth muscle cells. Cardiovasc. Res. 118, 622–637 (2022).
Scheppke, L. et al. Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basement membrane. Blood 119, 2149–2158 (2012).
Miyahara, S. et al. Alteration of leukocyte-endothelial cell interaction during aging in retinal microcirculation of hypertensive rats. Jpn. J. Ophthalmol. 50, 509–514 (2006).
Michaux, G. & Cutler, D. F. How to roll an endothelial cigar: the biogenesis of Weibel-Palade bodies. Traffic 5, 69–78 (2004).
Denis, C. V., André, P., Saffaripour, S. & Wagner, D. D. Defect in regulated secretion of P-selectin affects leukocyte recruitment in von Willebrand factor-deficient mice. Proc. Natl Acad. Sci. USA 98, 4072–4077 (2001).
Pendu, R. et al. P-selectin glycoprotein ligand 1 and beta2-integrins cooperate in the adhesion of leukocytes to von Willebrand factor. Blood 108, 3746–3752 (2006).
Bernardo, A. et al. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J. Thromb. Haemost. 3, 562–570 (2005).
Petri, B. et al. von Willebrand factor promotes leukocyte extravasation. Blood 116, 4712–4719 (2010).
Suidan, G. L. et al. Endothelial Von Willebrand factor promotes blood-brain barrier flexibility and provides protection from hypoxia and seizures in mice. Arterioscler Thromb Vasc Biol 33, 2112–2120 (2013).
Grässle, S. et al. von Willebrand factor directly interacts with DNA from neutrophil extracellular traps. Arterioscler. Thromb. Vasc. Biol. 34, 1382–1389 (2014).
Binet, F. & Cagnone, G. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 369, eaay5356 (2020).
Bettoni, S. et al. Interaction between multimeric von willebrand factor and complement: a fresh look to the pathophysiology of microvascular thrombosis. J. Immunol. 199, 1021–1040 (2017).
Hillgruber, C. et al. Blocking von Willebrand factor for treatment of cutaneous inflammation. J. Invest. Dermatol. 134, 77–86 (2014).
Wang, J. C. & Bennett, M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 111, 245–259 (2012).
Bian, J. et al. Relationship between serum FGF21 and vWF expression and carotid atherosclerosis in elderly patients with hypertension. J. Healthc. Eng. 2022, 6777771 (2022).
Sonneveld, M. A. et al. Von Willebrand factor in relation to coronary plaque characteristics and cardiovascular outcome. Results of the ATHEROREMO-IVUS study. Thromb. Haemost. 113, 577–584 (2015).
Bürrig, K. F. The endothelium of advanced arteriosclerotic plaques in humans. Arterioscler. Thromb. 11, 1678–1689 (1991).
Tohgi, H., Utsugisawa, K., Yoshimura, M., Nagane, Y. & Ukitsu, M. Local variation in expression of pro- and antithrombotic factors in vascular endothelium of human autopsy brain. Acta Neuropathol. 98, 111–118 (1999).
Bilora, F. et al. Do hemophilia A and von Willebrand disease protect against carotid atherosclerosis? A comparative study between coagulopathics and normal subjects by means of carotid echo-color Doppler scan. Clin. Appl. Thromb. Hemost. 5, 232–235 (1999).
Bilora, F., Boccioletti, V., Zanon, E., Petrobelli, F. & Girolami, A. Hemophilia A, von Willebrand disease, and atherosclerosis of abdominal aorta and leg arteries: factor VIII and von Willebrand factor defects appear to protect abdominal aorta and leg arteries from atherosclerosis. Clin. Appl. Thromb. Hemost. 7, 311–313 (2001).
Srámek, A., Reiber, J. H., Gerrits, W. B. & Rosendaal, F. R. Decreased coagulability has no clinically relevant effect on atherogenesis: observations in individuals with a hereditary bleeding tendency. Circulation 104, 762–767 (2001).
Srámek, A. et al. Patients with type 3 severe von Willebrand disease are not protected against atherosclerosis: results from a multicenter study in 47 patients. Circulation 109, 740–744 (2004).
Fuster, W. et al. Resistance to arteriosclerosis in pigs with von Willebrand’s disease. Spontaneous and high cholesterol diet-induced arteriosclerosis. J. Clin. Invest. 61, 722–730 (1978).
Methia, N., André, P., Denis, C. V., Economopoulos, M. & Wagner, D. D. Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood 98, 1424–1428 (2001).
Theilmeier, G. et al. Endothelial von Willebrand factor recruits platelets to atherosclerosis-prone sites in response to hypercholesterolemia. Blood 99, 4486–4493 (2002).
Kumari, M., Marmot, M. & Brunner, E. Social determinants of von willebrand factor: the Whitehall II study. Arterioscler. Thromb. Vasc. Biol. 20, 1842–1847 (2000).
Kim, J. et al. The effects of cadmium on VEGF-mediated angiogenesis in HUVECs. J. Appl. Toxicol. 32, 342–349 (2012).
Abu-Hayyeh, S., Sian, M., Jones, K. G., Manuel, A. & Powell, J. T. Cadmium accumulation in aortas of smokers. Arterioscler. Thromb. Vasc. Biol. 21, 863–867 (2001).
Shah, A. V., Birdsey, G. M. & Randi, A. M. Regulation of endothelial homeostasis, vascular development and angiogenesis by the transcription factor ERG. Vascul. Pharmacol. 86, 3–13 (2016).
Liu, J. et al. Vascular bed-specific regulation of the von Willebrand factor promoter in the heart and skeletal muscle. Blood 117, 342–351 (2011).
Miettinen, M. et al. ERG transcription factor as an immunohistochemical marker for vascular endothelial tumors and prostatic carcinoma. Am. J. Surg. Pathol. 35, 432–441 (2011).
McKay, K. M., Doyle, L. A., Lazar, A. J. & Hornick, J. L. Expression of ERG, an Ets family transcription factor, distinguishes cutaneous angiosarcoma from histological mimics. Histopathology 61, 989–991 (2012).
Minner, S. et al. High level of Ets-related gene expression has high specificity for prostate cancer: a tissue microarray study of 11 483 cancers. Histopathology 61, 445–453 (2012).
Haber, M. A. et al. ERG is a novel and reliable marker for endothelial cells in central nervous system tumors. Clin. Neuropathol. 34, 117–127 (2015).
O’Malley, D. P., Kim, Y. S. & Weiss, L. M. Distinctive immunohistochemical staining in littoral cell angioma using ERG and WT-1. Ann. Diagn. Pathol. 19, 143–145 (2015).
Vasuri, F. et al. ETS-related gene expression in healthy femoral arteries with focal calcifications. Front. Cell Dev. Biol. 9, 623782 (2021).
Birdsey, G. M. et al. The endothelial transcription factor ERG promotes vascular stability and growth through Wnt/β-catenin signaling. Dev. Cell 32, 82–96 (2015).
Moh-Moh-Aung, A. et al. Decreased miR-200b-3p in cancer cells leads to angiogenesis in HCC by enhancing endothelial ERG expression. Sci. Rep. 10, 10418 (2020).
Baltzinger, M., Mager-Heckel, A. M. & Remy, P. Xl erg: expression pattern and overexpression during development plead for a role in endothelial cell differentiation. Dev. Dyn. 216, 420–433 (1999).
Jahroudi, N. & Lynch, D. C. Endothelial-cell-specific regulation of von Willebrand factor gene expression. Mol. Cell Biol. 14, 999–1008 (1994).
Harvey, P. J., Keightley, A. M., Lam, Y. M., Cameron, C. & Lillicrap, D. A single nucleotide polymorphism at nucleotide -1793 in the von Willebrand factor (VWF) regulatory region is associated with plasma VWF:Ag levels. Br. J. Haematol. 109, 349–353 (2000).
Wang, X., Peng, Y., Ma, Y. & Jahroudi, N. Histone H1-like protein participates in endothelial cell-specific activation of the von Willebrand factor promoter. Blood 104, 1725–1732 (2004).
Peng, Y. & Jahroudi, N. The NFY transcription factor functions as a repressor and activator of the von Willebrand factor promoter. Blood 99, 2408–2417 (2002).
Jahroudi, N., Ardekani, A. M. & Greenberger, J. S. An NF1-like protein functions as a repressor of the von Willebrand factor promoter. J. Biol. Chem. 271, 21413–21421 (1996).
Waisberg, M., Joseph, P., Hale, B. & Beyersmann, D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology 192, 95–117 (2003).
ATSDR. ATSDR’s substance priority list. https://www.atsdr.cdc.gov/spl/ (2019).
Satarug, S., Vesey, D. A. & Gobe, G. C. Current health risk assessment practice for dietary cadmium: Data from different countries. Food Chem. Toxicol. 106, 430–445 (2017).
Zalups, R. K. & Ahmad, S. Molecular handling of cadmium in transporting epithelia. Toxicol. Appl. Pharmacol. 186, 163–188 (2003).
ATSDR. Case Studies in Environmental Medicine (CSEM) Cadmium Toxicity. https://www.atsdr.cdc.gov/csem/cadmium/docs/cadmium.pdf (2010).
Tinkov, A. A. et al. Cadmium and atherosclerosis: a review of toxicological mechanisms and a meta-analysis of epidemiologic studies. Environ. Res. 162, 240–260 (2018).
Diaz, D. et al. Low-level cadmium exposure and atherosclerosis. Curr. Environ. Health Rep. 8, 42–53 (2021).
Tellez-Plaza, M. et al. Cadmium exposure and all-cause and cardiovascular mortality in the U.S. general population. Environ. Health Perspect. 120, 1017–1022 (2012).
Messner, B. et al. Cadmium is a novel and independent risk factor for early atherosclerosis mechanisms and in vivo relevance. Arterioscler. Thromb. Vasc. Biol. 29, 1392–1398 (2009).
Subramanyam, G., Bhaskar, M. & Govindappa, S. The role of cadmium in induction of atherosclerosis in rabbits. Indian Heart J. 44, 177–180 (1992).
Nai, G. A., Golghetto, J. J., Estrella, M. P., Alves, J. A. & Garcia, L. A. pH dependence of cadmium-contaminated drinking water on the development of cardiovascular injury in Wistar rats. Biol. Trace Elem. Res. 165, 81–85 (2015).
Revis, N. W., Zinsmeister, A. R. & Bull, R. Atherosclerosis and hypertension induction by lead and cadmium ions: an effect prevented by calcium ion. Proc. Natl Acad. Sci. USA 78, 6494–6498 (1981).
Revis, N. W., Major, T. C. & Horton, C. Y. The effects of calcium, magnesium, lead, or cadmium on lipoprotein metabolism and atherosclerosis in the pigeon. J. Environ. Pathol. Toxicol. 4, 293–303 (1980).
Knoflach, M. et al. Non-toxic cadmium concentrations induce vascular inflammation and promote atherosclerosis. Circ. J. 75, 2491–2495 (2011).
Oliveira, T. F. et al. Chronic cadmium exposure accelerates the development of atherosclerosis and induces vascular dysfunction in the aorta of ApoE(-/-) Mice. Biol. Trace Elem. Res. 187, 163–171 (2019).
Li, Y. et al. Plasma von Willebrand factor level is transiently elevated in a rat model of acute myocardial infarction. Exp. Ther. Med. 10, 1743–1749 (2015).
Romero, L. I., Zhang, D. N., Herron, G. S. & Karasek, M. A. Interleukin-1 induces major phenotypic changes in human skin microvascular endothelial cells. J. Cell Physiol. 173, 84–92 (1997).
Cai, H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc. Res. 68, 26–36 (2005).
Acknowledgements
This study was supported by grants from the National Nature Science Foundation of China (82241030, 82171318, and 82011530024), the State Program of Scientific Research “Natural Resources and the Environment” (3.01, 2020-2021, Belarus), Academic Promotion Program of Shandong First Medical University (2019QL014) and Shandong Taishan Scholarship (Ju Liu).
Author information
Authors and Affiliations
Contributions
X.W. wrote the initial draft of the review and prepare the figure; M.N.S. and C.M.K. revised the manuscript; J.L. conceived and revised the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Starodubtseva, M.N., Kapron, C.M. et al. Cadmium, von Willebrand factor and vascular aging. npj Aging 9, 11 (2023). https://doi.org/10.1038/s41514-023-00107-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41514-023-00107-3
