
Abstract
Piezoelectricity of organic polymers has attracted increasing interest because of several advantages they exhibit over traditional inorganic ceramics. While most organic piezoelectrics rely on the presence of intrinsic local dipoles, a highly nonlocal electronic polarisation can be foreseen in conjugated polymers, characterised by delocalised and highly responsive π-electrons. These 1D systems represent a physical realisation of a Thouless pump, a mechanism of adiabatic charge transport of a topological nature which results, as shown in this work, in anomalously large dynamical effective charges, inversely proportional to the bandgap energy. A structural (ferroelectric) phase transition further contributes to an enhancement of the piezoelectric response reminiscent of that observed in piezoelectric perovskites close to morphotropic phase boundaries. First-principles density functional theory (DFT) calculations performed in two representative conjugated polymers using hybrid functionals, show that state-of-the-art organic piezoelectric are outperformed by piezoelectric conjugated polymers, mostly thanks to strongly anomalous effective charges of carbon, larger than 5e—ordinary values being of the order of 1e—and reaching the giant value of 30e for band gaps of the order of 1 eV.
Similar content being viewed by others


Introduction
Piezoelectricity is a well-known phenomenon characterising those materials which have the property to generate a surface charge, and hence an electric tension, when subject to a stress, or conversely to deform elastically in response to an external electric field. Thanks to the possibility they offer to convert mechanical energy into electrical energy and vice-versa, piezoelectric materials are of great interest in various fields and for many applications, from macro- to microscopic electromechanical devices, to energy harvesting and much more1,2. The most widely used piezoelectric materials are inorganic perovskites, such as lead zirconate titanate (PZT), for their high electromechanical response3,4,5. However, inorganic materials have very low mechanical flexibility, high fabrication costs and often are toxic because of the lead they contain. These facts motivated an intense research activity aimed at developing and identifying lead-free piezoelectric ceramics5,6. A promising alternative is to exploit piezoelectric properties of organic materials, a route that has been trodden with some success since the early 1990s, mostly focusing on the wide class of organic polymers displaying high flexibility, low fabrication costs and bio-compatibility7,8,9,10. In this context, the most studied organic piezoelectric is polyvinylidene fluoride (PVDF)8,11, a saturated polymer derived from polyethylene and comprising molecular units with net (electrical) dipole moments, thus giving rise to ferroelectricity of conformational origin due to the rotation of chains’ segments from non-polar to polar isomers.
Despite the intense research efforts and the progress made in the field of organic piezoelectrics, to date, inorganic ceramics still display much better piezoelectric performance than organic counterparts. The piezoelectric response in PZT and related inorganic materials is strongly enhanced at morphotropic phase boundaries (MPB), marking a composition-driven structural transition between two competing, nearly energetically degenerate phases with distinct symmetries12,13,14. On general grounds, the properties enhancement close to such phase transition may be traced back to the flattening of free-energy surfaces, easing polarisation extension and/or rotation, as extensively discussed and observed mostly in perovskite oxides15. Recently, the concept of MPB has been loosely extended to the family of P(VDF-TrFE) copolymers16,17. Here, the introduction of different TrFE monomers in the semicrystalline PVDF structure has been proposed to lead to an enhanced conformational competition reminiscent of the structural competition realised at MPB, further suggesting an optimal chemical composition for maximising the piezoelectric coefficient. Even though at the optimal 'morphotropic' composition, the piezoelectric coefficient roughly doubles the typical values of PVDF, it is still smaller by one order of magnitude compared to characteristic piezoelectric coefficients of inorganic oxide ceramics such as PZT.
Besides saturated polymers as PVDF and P(VDF-TrFE), whose ferroelectric and piezoelectric properties rely on the ordering of built-in molecular dipoles and, as such, require appropriate poling treatments, a natural alternative choice is represented by conjugated polymers, such as archetypical polyacetylene (PA). Characterised by a delocalised π−orbital along their backbone, these polymers are widely studied for their peculiar electronic properties18,19. If specific symmetry-lowering effects allowing for piezoelectricity and ferroelectricity are met in a conjugated polymer, one may expect a large polar response of electronic origin due to the redistribution of the responsive π−electronic density along the polymer’s backbone. Because of the delocalised nature of conjugated π−state (of Bloch-type in an infinite periodic chain), small changes of atomic positions may lead to a global shift of electrons, revealing the strong nonlocal and ultimately topological character of electronic polarisation in these quasi-1D systems20. Indeed, conjugated polymers have been theoretically put forward as a potential new class of “electronic ferroelectrics”21.
In order to explore the potential piezoelectricity of conjugated polymers, we adopt the simple model originally introduced by Rice and Mele to study soliton excitations of linearly conjugated diatomic polymers22, later become a prototypical model for 1D ferroelectricity20,23 and Thouless adiabatic pumping24,25. Building on these well-established results, we study how the interplay between these mechanisms affects the system’s response to a mechanical strain. We find that polar responses, such as Born effective-charge tensors—also known as atomic polar tensors—and piezoelectric coefficients, can be strongly enhanced close to the dimerisation phase transition characterised by a bond-length alternation (BLA) between neighbouring atoms along the chain. This transition can be controlled by changes in the chemical composition of the polymers or by tuning the electron–phonon (e–ph) interaction, that is expected to be sensitive to the dielectric environment of individual chains. The strength of the piezoelectric effect is found to be determined partly by the MPB-like enhancement—relying on the second-order nature of the transition—but mostly by a giant effective charge, ultimately due to the topological nature of Thouless’ adiabatic charge transport in this class of materials. We verified the model-based predictions and theoretical picture by performing density functional theory (DFT) calculations using a range-separated hybrid functional on two prototypical conjugated polymers derived from polyacetylene, confirming strong enhancement of effective charges and consequently of piezoelectric response, that is found to be larger than the one observed in the celebrated organic piezoelectric PVDF for a wide range of parameters.
Results
A model for piezoelectric conjugated polymers
To study the effects of strain on conjugated polymers, we generalise the Rice-Mele model22 of a 1D linear chain with a unit cell of length a0 containing two atoms. The only possible strains for a 1D infinite periodic chain are contraction/dilatation of the unit cell, defined by a(ϵ) = a0(1 + ϵ), where the adimensional parameter ϵ quantifies the strain along the direction of the chain. In the nearest neighbours tight-binding approximation, the modulation of the hopping energy between atoms at site i and i + 1 can be modelled at linear order in atoms’ displacement and strain as ti,i+1 = t(ϵ) + (−1)i+1δt(ϵ), with
Equation (1) accounts for the effect of strain on the hopping energy t0 between equidistant atoms, while Equation (2) describes the variation with respect to t(ϵ) caused by atoms’ displacement; at the lowest order, we can assume the same adimensional e–ph coupling parameter β > 0 in describing both effects. The term u(ϵ) is an adimensional fractional coordinate measuring the relative displacement of the two atoms in the unit cell, as defined in the Methods. We indicate with the term Δ ≥ 0 the onsite energy difference between neighbours. If Δ = 0, the atoms in the unit cell are equivalent, and we recover the well-known SSH model26, used to describe polyacetylene. However, in order to have a non-trivial polar response, it is necessary to break atoms’ equivalence (Δ ≠ 0) as, e.g., in substituted polyacetylenes (SPA)27, a class of conjugated polymers formed by inequivalent monomers which can be obtained substituting (one of) the atoms in the C2H2 unit of PA with some element(s) or compound(s). A representation of the model is in the insets of Fig. 1a, b. For simplicity, we consider only longitudinal displacements, parallel to the linear-chain direction. As we are interested in the equilibrium structures at T = 0 K, we define the optimal displacement \(\overline{u}\) as the one which minimises the total energy, given a set of material-dependent parameters and a strain. For more information on the model and the derivation of the above quantities, see the Methods and the Supplementary Methods.

a, b Structural phase transition with order parameter \(\overline{u}\) as a function of the onsite energy difference Δ and of the e–ph coupling β. As shown in Supplementary Fig. 2, when Δ < Δc (β > βc) the total energy Etot(u) has a double-well profile with two minima at \(| \overline{u}|\, \ne \,0\), resulting in a distorted chain with bond-length alternation. When Δ > Δc (β < βc) the minimum of Etot(u) is at \(\overline{u}=0\) and the atoms become equidistant. The behaviour of the order parameter is typical of second-order phase transitions, being \(\overline{u}\propto | \Delta -{\Delta }_{{{{\rm{c}}}}}{| }^{1/2}\) (\(\overline{u}\propto | \beta -{\beta }_{{{{\rm{c}}}}}{| }^{1/2}\)), and the system remains insulating in both phases with gap \({E}_{{{{\rm{gap}}}}}=2\sqrt{{\Delta }^{2}+4{(\beta {t}_{0}\overline{u})}^{2}}\). c, d Internal strain and effective charge across the phase transition. The inclusion of strain ϵ in the model affects the critical value Δc(ϵ) (βc(ϵ)) and, as expected in a second-order transition, the internal strain displays a diverging behaviour close to the critical point, \(\partial \overline{u}/\partial \epsilon \propto | \Delta -{\Delta }_{{{{\rm{c}}}}}{| }^{-1/2}\) (\(\partial \overline{u}/\partial \epsilon \propto | \beta -{\beta }_{{{{\rm{c}}}}}{| }^{-1/2}\)). Near the critical point, a huge polar response is also present, quantified by the effective charge \({Z}^{* }\propto \beta \Delta /{E}_{{{{\rm{gap}}}}}^{2}\) (see Methods). Approaching the critical point from the undimerized phase, Z* is inversely proportional to the gap \({E}_{{{{\rm{gap}}}}}(\overline{u}=0)\equiv \Delta\) (c) and linear in β (d). Since the gap is constant as a function of Δ and linear in β when approaching the critical point from the dimerised phase, Z* displays a linear behaviour in Δ (c), and it is inversely proportional to β (d). e, f The piezoelectric coefficient diverges when approaching the critical point from the dimerised phase. As shown in the inset, the major contribution is due to the internal-relaxation term of Equation (5). The topological nature of the enhancement guarantees its stability.
In the absence of strain (ϵ = 0), the properties of the model are well known. In the SSH model (Δ = 0), a finite \(\overline{u}\,\ne\, 0\) is allowed by an infinitely small e–ph interaction, conjuring with a Fermi-surface nesting and Peierls electronic instability to produce a dimerised phase with bond-length alternation and the opening of a gap in the energy spectrum \({E}_{{{{\rm{gap}}}}}=4| \delta t| =4\beta {t}_{0}\overline{u}\). Breaking the equivalence between atoms (Δ ≠ 0) also opens a gap which is expected to counteract the Peierls instability and the related bond dimerisation. The lower-symmetry structure can be stabilised only by a finite and strong enough e–ph interaction, resulting in a gap \({E}_{{{{\rm{gap}}}}}=2\sqrt{{\Delta }^{2}+4\delta {t}^{2}}=2\sqrt{{\Delta }^{2}+4{(\beta {t}_{0}\overline{u})}^{2}}\). Indeed, as shown in Fig. 1a, increasing Δ at fixed e–ph coupling induces a second-order structural phase transition at a critical value Δc with order parameter \(\overline{u}\) between a distorted (\(\overline{u}\,\ne\, 0\)) and a higher-symmetric undimerized (\(\overline{u}=0\)) phase, both displaying an insulating character with a finite bandgap (see also Supplementary Fig. 1 for the band structure of the system and Supplementary Methods for more details). Such structural transition from the dimerised to undimerized phase can also be induced by decreasing the e–ph coupling at fixed non-zero onsite energy difference, as discussed in the next section and displayed in Figure b. In a 2D parametric (Δ, δt)-space, where δt depends—through optimal \(\overline{u}\)—on both Δ and β, the origin of the axes corresponds to a metallic system while the rest of the space to an insulator. Since the milestone paper by Vanderbilt and King-Smith24, it is known that if this system undergoes an adiabatic evolution along a loop enclosing the origin of this space, a quantised charge ∣e∣ is pumped out. This is a prototypical example of the adiabatic charge transport known as Thouless’ pump25,28 realised e.g. in ultracold fermions29. The crucial ingredient is the presence of the metallic point in the domain enclosed by the loop and, in this sense, it is an example of a topological phenomenon (see Supplementary Methods). Previous studies on low-dimensional systems showed how topology may reflect onto polar responses, resulting, e.g., in electronic polarisation in quasi-1D systems20 or in large effective charges and piezoelectric coefficients directly related to valley Chern numbers in 2D systems30,31. In the next two Sections, we use the generalised Rice-Mele model to show how the Thouless-pump topological mechanism conjures with the bond dimerisation to give raise to an enhanced piezoelectric effect, and we discuss how the latter can be tuned by acting on the two independent parameters Δ and β.
Morphotropic-like enhancement of the piezoelectric response
In order to have a non-trivial piezoelectric response, the chain must not have points of inversion symmetry, a requirement that is met when the equivalence between atoms is broken in a distorted chain, i.e. both Δ ≠ 0 and \(\overline{u}\,\ne\, 0\). In this case, the chain becomes also ferroelectric with a net dipole moment per unit cell P23. The electromechanical response is quantified by the piezoelectric coefficient cpiezo, defined as the variation of P due to an applied homogeneous strain ϵ, namely
where the derivative of P is decomposed in two contributions. The first one is the so-called clamped ions term
which is obtained keeping fixed the relative position of the ions in the unit cell, i.e. for fixed internal fractional coordinate \({\overline{u}}_{0}=\overline{u}(0)\). It can be shown (see Methods) that \(| {c}_{{{{\rm{piezo}}}}}^{{{{\rm{c.i.}}}}}| \le | e| \beta /2\pi\). The second term of Equation (3) takes into account the effect of strain on the internal coordinate \(\overline{u}(\epsilon )\) and defines the internal-relaxation contribution
where we defined the effective charge Z*, namely a measure of how rigidly the electronic charge distribution follows the displacement of the nuclei, as
The inclusion of strain in the Rice-Mele model affects explicitly the critical value Δc(ϵ) of the phase transition. The behaviour of \(\overline{u}(\epsilon )\) as Δ approaches Δc(ϵ) is shown in Fig. 1a, following the expected behaviour \(\overline{u}(\epsilon )\propto | \Delta -{\Delta }_{{{{\rm{c}}}}}(\epsilon ){| }^{1/2}\) of the order parameter of second-order phase transitions (see Supplementary Methods). It thus follows that:
Equation (7) implies that the internal-relaxation term diverges as we approach the critical point Δc(ϵ) from the distorted phase, in analogy with the MPB mechanism at play in some ferroelectric oxides. Indeed, as the Δ parameter of the Rice-Mele model accounts for the composition of the system, it allows to continuously tune a morphotropic-like phase transition from the distorted phase (lower symmetry, ferroelectric) to the undistorted one (higher symmetry, paraelectric). On the other hand, at a fixed Δ ≠ 0 suppressing the Peierls electronic instability, the second-order phase transition can be driven by the e–ph coupling, as shown in Fig. 1b, allowing one to define a critical value βc separating the undimerized and dimerised phases (see Supplementary Methods). Clearly, the order parameter as a function of the variable driving the transition would still follow the expected behaviour \(\overline{u}(\epsilon )\propto | \beta -{\beta }_{{{{\rm{c}}}}}(\epsilon ){| }^{1/2}\), implying:
i.e. a diverging internal strain when approaching the critical point from the dimerised phase.
Topological contribution to the enhancement
In principle, the diverging behaviour of the internal strain, Equation (7) or (8), guarantees the existence of piezoelectric polymers with arbitrarily high response when close to a morphotropic-like phase boundary, irrespective of the prefactor, namely the effective charge Z*. However, this specific enhancement is a consequence of the second-order transition. Numerical evidence in linear acetylenic carbon chains32 show that quantum anharmonic effects (QAE) may change the order of the structural phase transition, therefore damping the diverging behaviour of \({c}_{{{{\rm{piezo}}}}}^{{{{\rm{i.r.}}}}}\). A robust enhancement of the piezoelectric coefficient against QAE would depend, therefore, on the strength of the polar response embodied by Z*. Measuring how the electronic charge distribution follows the displacement of the nuclei, effective charge’s behaviour in this system is strictly related to the topological charge transport of the Thouless pump28. A simple geometric argument shows that indeed the effective charge Z* diverges as 1/Egap in the undimerized phase (see Methods and Supplementary Methods). As first noted in ref. 20, such a remarkable behaviour is a consequence of the topology of the domain of the dipole moment P. As Egap goes to 0, we get closer to the metallic point, where P is not well defined, and even an infinitesimal atomic displacement causes a huge redistribution of the charge density. We contrast this result with the predicted behaviour of polar responses in 2D gapped graphene, where both piezoelectric coefficient and effective charges were found to be independent on the bandgap amplitude31. Indeed, electron-strain/lattice couplings in 2D hexagonal crystals can be described as gauge fields30,31 whose effect is to shift the Dirac cone of an amount proportional to the coupling constants, causing the latter to be the only relevant quantities determining the strength of polar responses. In the 1D chain, instead, the e–ph interaction contributes, through dimerisation, to the gap opening, thus directly affecting the Thouless-pump topological enhancement of effective charge. We further remark that the absence of a structural transition in gapped graphene causes the piezoelectric response to be mostly due to the clamped-ion contribution, the internal-relaxation one contributing roughly 25% to the total response31.
The evolution of Z* as a function of Δ/Δc and β/βc is shown in Fig. 1c, d. Even though the system always displays a finite gap preventing the metallic divergence of the effective charge, Z* reaches the giant value of ~30∣e∣ at the critical points. Such anomalously large effective charges can not be ascribed to the mixed covalent-ionic character of the system only: it can be shown (see Supplementary Discussion and Supplementary Fig. 8 for the simple case of a heteropolar biatomic molecule) that tuning the bond character can lead to a finite enhancement of the effective charges, which are typically only few times the value of the nominal charge33. Unlike the MPB-related enhancement of the internal strain, also shown in Fig. 1c, d, the topological behaviour is expected to be much more stable with respect to QAE, guaranteeing the enhancement of the electromechanical response. The total piezoelectric coefficient, comprising both the clamped ion and internal-relaxation contributions, is shown in Fig. 1e, f as a function of parameters Δ/Δc and β/βc. Insets highlight how the piezoelectric coefficient is mostly contributed by the internal-relaxation contribution, that is strongly enhanced by the combined effect of diverging internal strain \(\partial \bar{u}/\partial \epsilon\) and anomalously large effective charges. We remark the importance of both mechanisms, since anomalous effective charges alone, in general, do not guarantee piezoelectric effects if inversion symmetry is kept, as in centrosymmetric CaTiO3 and SrTiO333, or if the internal strains are small, as in 2D hexagonal systems and gapped graphene31.
Numerical calculations
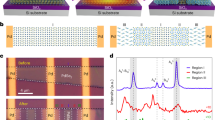
We performed ab initio calculations in the framework of density functional theory (DFT) to validate our model predictions, choosing two conjugate polymers representative of the broad class of SPA. One is monofluorinated polyacetylene (MFPA) made by the repetition of the unit CH-CF, obtained by substituting one hydrogen atom of the C2H2 unit of PA with fluorine. The other is polymethineimine (PMI), obtained substituting a CH pair with a nitrogen atom to obtain the unit N-CH. For simplicity, we considered the all-trans structures shown in Fig. 2a, b, whose fundamental physical properties are captured by the Rice-Mele model. Even though controlling the fraction of substituted atoms may, in principle, induce a morphotropic-like transition, this approach poses many challenges both from the computational and experimental side: to our specific purposes, it wouldn’t allow us to study the phase transition and the associated predicted enhancement of piezoelectric effect by varying with continuity an external parameter (as Δ in the model). As discussed in the previous sections, the internal-strain and the effective-charge enhancements may be also induced by tuning the parameter β. To achieve this computational task, we take advantage of the effect of screened-Coulomb vertex corrections to the dressing of the e–ph coupling34, leading to an enhancement of e–ph itself especially strong in low-dimensional materials and when phonons at zone boundary are involved35,36,37,38. Such screened-Coulomb-mediated e–ph enhancement can be captured by hybrid functionals incorporating a fraction of the exact exchange36,39. Its inclusion has been proven essential for describing the bond-length alternation of trans-polyacetylene40,41 and related 1D polymers32,42, whose BLA is typically underestimated by standard local-density or generalised-gradient approximations, indirectly pointing to an enhancement of the e–ph coupling due to electron–electron interaction. Range-separated hybrid (RSH) functionals represent an ideal choice for our purpose, as they have been designed to better account for the screened-Coulomb vertex corrections. The latter can be effectively tuned by acting on the long-range (LR) mixing parameter cLR that accounts for the fraction of LR exact exchange in RSH functionals, thus providing a computational knob to continuously vary β. We further remark that the strength of e–ph enhancement due to screened-Coulomb effects can be ideally controlled by modifying the screening itself, as proposed for doped graphene43. Since the optimal mixing parameter cLR is inversely proportional to the scalar dielectric constant of the environment in order to enforce the correct asymptotic potential44,45,46,47,48, we speculate that controlling the dielectric environment may represent a viable strategy, alternative and complementary to controlling the fraction of substituted atoms, for tuning and optimising the piezoelectric response of conjugated polymers.

a Monofluorinated polyacetylene (MFPA). b Polymethineimine (PMI). In c, d for MFPA and PMI, respectively, is shown the behaviour of the internal coordinate \({\overline{u}}_{0}\) for different values of the long-range mixing parameter cLR put in the range-separated xc-functional in the DFT calculations. Consistently with the prediction of the model, we observe the behaviour \(\overline{u}\simeq | {c}_{{{{\rm{LR}}}}}-{\overline{c}}_{{{{\rm{LR}}}}}{| }^{1/2}\), with \({\overline{c}}_{{{{\rm{LR}}}}}^{{{{\rm{MFPA}}}}}\simeq 14 \%\) and \({\overline{c}}_{{{{\rm{LR}}}}}^{{{{\rm{PMI}}}}}\simeq 26 \%\). In e, f, for MFPA and PMI respectively, the behaviour of the \(\partial {\overline{u}}_{0}/\partial \epsilon\) is reported, along with the values of the effective charge Z*. For each polymer, we chose \({Z}^{* }={Z}_{{{{\rm{C}}}},xx}^{* }\) (for MFPA the C bound to the F). In agreement with the model, on the one hand we observe a further hint of the morphotropic-like nature of the transition while on the other hand the huge values of Z* stands out, in particular in the region near the critical points. In g, f is shown how the piezoelectric coefficient is greatly enhanced when reaching the critical points from the less symmetric phase. The comparison between \({c}_{{{{\rm{piezo}}}}}^{{{{\rm{c.i.}}}}}\) and \({c}_{{{{\rm{piezo}}}}}^{{{{\rm{i.r.}}}}}\), in the insets, highlights the internal-relaxation origin of the enhancement. Furthermore, the comparison with the results obtained putting the values of e, f in Equation (5) shows that the model very well describes the nature of the enhancement.
Motivated by these reasons, we performed structural optimisation of both MFPA and PMI for different values of the LR mixing parameter cLR. For consistency with the model, we considered the coordinates of the C and N atoms along the principal axis of the chain, which we take as the x-axis, to compute the internal coordinate. More details on the effect of cLR on polymers’ structures are provided in the Supplementary Methods. The evolution of \({\overline{u}}_{0}\) displayed in Fig. 2c, d, clearly hints at the presence of a second-order phase transition triggered by cLR for both polymers: the dimerised phase is suppressed by lowering the fraction of mixing, the order parameter showing the expected behaviour as it approaches the second-order phase-transition critical point, in excellent qualitative agreement with model results shown in Fig. 1b, d, f and confirming a posteriori the direct proportionality between cLR and β. The second-order character of the phase transition is further confirmed by the softening of the corresponding optical phonon found in the higher-symmetry phase when increasing cLR (Supplementary Fig. 5), signalling the onset of a dynamical instability of the undimerized structure.
In Fig. 2e, f the behaviour of Z* and of \(\partial \overline{u}/\partial \epsilon\) calculated from first principles is shown. For MFPA, we took \({Z}^{* }={Z}_{{{{{\rm{C}}}}}_{{{{\rm{F}}}}},xx}^{* }\) where CF is the carbon atom bound to the fluorine, while for PMI \({Z}^{* }={Z}_{{{{\rm{C}}}},xx}^{* }\). The full tensors of the effective charges of all the atoms are reported in Supplementary Tables I, II, and III. We highlight the qualitative agreement with the prediction of the model, in particular the huge enhancement of the effective charges around the critical point \({\overline{c}}_{{{{\rm{LR}}}}}\), reaching the strongly anomalous values of ~30∣e∣ and ~15∣e∣ in correspondence of \({\overline{c}}_{{{{\rm{LR}}}}}\) for MFPA and PMI, respectively. The covalent character of bonds along the chain prevents a precise definition of the nominal reference value for C, that can be, however, assumed to be of the order of 1∣e∣, as the nominal ionic charges for H and F are respectively +1∣e∣ and −1∣e∣. Effective charges of carbon in both considered chains are strongly anomalous for all considered long-range mixing parameters, displaying values between 5∣e∣ and 30∣e∣ even for band gaps exceeding 6 eV. These anomalous values exceed even those reported in oxide ferroelectrics, where effective charges are typically two or three times larger than nominal reference values33. Figure 2g, h display the behaviour of the piezoelectric coefficients computed ab initio, taking into account also the effects of transverse displacements. In the insets, the different contributions \({c}_{{{{\rm{piezo}}}}}^{{{{\rm{i.r.}}}}}\) and \({c}_{{{{\rm{piezo}}}}}^{{{{\rm{c.i.}}}}}\) are compared, highlighting the internal-relaxation origin of the enhancement. Using the ab initio values of Z* and \(\partial \overline{u}/\partial \epsilon\) of Fig. 2e, f, we compare the values of \({c}_{{{{\rm{piezo}}}}}^{{{{\rm{i.r.}}}}}\) computed without approximations with those obtained using Equation (5) of the model. The agreement between the two approaches is both qualitatively and quantitatively excellent, notwithstanding the simplifying description provided by the Rice-Mele model, that neglects structural details specific to the two considered polymers as well as transverse displacements. We highlight that despite the behaviour \(\partial \overline{u}/\partial \epsilon \propto | {c}_{{{{\rm{LR}}}}}-{\overline{c}}_{{{{\rm{LR}}}}}{| }^{-1/2}\), the main contribution to the piezoelectric coefficient is given by the effective charges. The large values attained in a finite range around the second-order critical point and their ultimately topological origin suggest that the piezoelectric effect is robust against quantum and anharmonic effects that may change the order of the phase transition, as predicted in carbyne32.
Discussion
Finally, we compare the results for the piezoelectric coefficients of MFPA and PMI with those of the best and most widely used piezoelectric PVDF polymer, made by the repetition of the unit CF2-CH2. The Rice-Mele model fails to capture its main properties, this polymer being not conjugated. Its piezoelectricity indeed derives from the presence of a net dipole moment transverse to the chain, whereas the electromechanical response predicted in conjugated polymers is longitudinal to the chain and ultimately due to the topological-morphotropic enhancement. To have a consistent comparison with available experimental data for PVDF, we computed ab initio the converse piezoelectric coefficient dpiezo, which measures the response with respect to an external stress, rather than a strain (see Methods). The results for the converse piezoelectric coefficients of PVDF are compared with those of MFPA and PMI in Fig. 3 and are consistent with the values computed in ref. 49. Even though the calculated dpiezo is smaller than reported experimental values, a direct comparison to experiments is hardly drawn because, e.g. of the polymorphic character or low crystallinity of experimental samples, as noticed also in ref. 49. We remark that the piezoelectric response in PVDF is found to be independent of the fraction of exact exchange, confirming the utterly different nature of the electromechanical response in such a non-conjugated polymer. We finally mention that mildly anomalous effective charges have been also reported for PVDF50, the carbon effective charge, however not exceeding 1.5∣e∣, consistently with our results provided in Supplementary Table III. On the other hand, both MFPA and PMI display a rather large range of values that are larger than calculated dpiezo of PVDF, with up to a six-fold enhancement for PMI close to the dimerisation point. The robustness of the enhancement mechanism is confirmed also by the comparison with the piezoelectric coefficients calculated in packed MFPA chains, as shown in Supplementary Fig. 9. Even though the selected prototypical SPAs may not be the most efficient ones for practical realisation and engineering of their functional properties, the comparison with first-principles estimate of one of the best available piezoelectric polymer alongside the general validity of the proposed model and consequent robustness of its electromechanical response put forward the broad class of π-conjugated polymers as a promising field for organic piezoelectrics with enhanced functionalities. Additionally, its inverse proportionality to the bandgap provides a possible material-design principle for driving the quest of organic polymers with enhanced piezoelectric response.

Comparison of the converse piezoelectric coefficients of MFPA and PMI with respect to PVDF, the current best and most widely used organic piezoelectric. In principle, the mechanism of the morphotropic-topological enhancement allows conjugated polymers to outperform the state-of-the-art.
Methods
Details on the model
The Hamiltonian of the chain is the sum of two terms Htot = HL + He. The lattice contribution HL accounts for the displacement of the atoms with respect to their position in a uniformly spaced chain:
where mi and δri are mass and displacement of atom i, pi its momentum, and K is a spring constant term. In the nearest neighbours tight-binding approximation, the electronic contribution He reads
where the factor 2 accounts for the spin degeneracy, \({c}_{i}^{{\dagger} }\)/ci are creation/annihilation operators for electrons, ti,i+1 is a hopping energy while Δ > 0 accounts for the onsite energy difference between neighbours. The adimensional order parameter u(ϵ) measures the relative displacement between neighbours and is defined as
The total energy of the system Etot(u) as a function of the order parameter u has two contributions, namely
The first term EL(u) accounts for lattice distortion and is quadratic in u, whereas the electronic contribution Ee(u) is linear in u. For a given set of material-dependent parameter t0, K, Δ, β, and the strain ϵ, we find the optimal displacement \(\overline{u}\)—defined as the one which minimises the total energy—as shown in detail in the Supplementary Methods.
We computed the dipole moment per unit cell P using the Berry-phase approach51,52. In the limit Egap ≪ t(ϵ) it holds (see Supplementary Methods)
with \(\tan \theta (\epsilon ,\overline{u}(\epsilon ))=\Delta /2\delta t(\epsilon ,\overline{u}(\epsilon ))\), where we emphasise that the dependence on the strain enters both directly and through \(\overline{u}(\epsilon )\). Using Equation (2) and (13) we have
where we notice that \(| {c}_{{{{\rm{piezo}}}}}^{{{{\rm{c.i.}}}}}| \le | e| \beta /2\pi\), the maximum achievable value being directly proportional to the e–ph coupling constant β.
Plugging Equation (2) in Equation (13), we obtain an explicit expression for the effective charge in both the dimerised and undimerized phases:
where \(\sin \theta =2\Delta /{E}_{{{{\rm{gap}}}}}\). In the undimerized phase \(\overline{u}=0\) implies \(\sin \theta =1\), hence Z* ~ β/Egap ~ β/Δ. The diverging behaviour can be further understood using a simple geometric argument that highlights its topological origin. From Equation (13), polarisation is proportional to an angle θ spanning the parametric (Δ, δt)-space along a circumference with radius Egap. As δt ∝ u, it follows that in the undimerized phase Egapdθ ∝ du, hence Z* = ∂P/∂u ∝ ∂θ/∂u ∝ 1/Egap (more details in the Supplementary Methods and Supplementary Fig. 3).
The parameters used to produce the results in Fig. 1 were obtained fitting the model with the PBE053 total energy profile of carbyne, an infinite-length straight chain of carbon atoms, as a function of the relative displacement of the two carbon atoms of its unit cell. In particular, with the value a0 = 2.534 Å obtained through a cell-relaxation procedure, the fit yields t0 = 2.239 eV, β = 3.906 and K = 127.9 eV/Å2, whereas for carbyne it holds Δ = 0. The behaviour with respect to β/βc was obtained fixing a finite Δ = 1.05Δc. We highlight that each carbon atom of carbyne contributes with two electrons to the π-orbital, so it is necessary to put an additional factor of 2 in front of Equation (10) to account for this degeneracy.
Computational details
All DFT calculations were performed using CRYSTAL code54,55, which employs a basis of local Gaussian-type functions. This approach allows for the simulation of truly isolated systems, as the 1D polymers addressed in our work, using the hybrid functionals, proven to be essential for accurately reproducing the physics of 1D chains,32,41. The Gaussian-type basis set significantly reduces the computational cost of evaluating real-space integrals and, hence, it allows to drastically reduce the computational cost when compared, e.g., with state-of-the-art plane-waves based DFT codes. We used a triple-ζ-polarised Gaussian-type basis56 with real-space integration tolerances of 10-10-10-15-30 and an energy tolerance of 10−10 Ha for the total energy convergence. We customised a range-separated LC-ωPBE hybrid exchange-correlation functional57 varying the value of the long-range (LR) mixing parameter cLR which enters in the definition of the LR part of the functional, namely
When cLR = 0, the PBE functional is recovered, while if cLR = 1, we have a pure Hartree-Fock (HF) exchange. The long-range terms in round brackets depend on the range-separation parameter ω that enters in the decomposition of the Coulomb operator 1/r as
where \({{{\rm{erf}}}}(\cdot )\) is the error function; the first and second term in the right-hand side of Eq. (17) account for the short- and long-range part of the Coulomb operator, respectively. All the presented values were obtained with ω = 0.4 a\({}_{0}^{-1}\). For each cLR, a geometric optimisation was performed, and all quantities were computed on the equilibrium configurations (see Supplementary Figs. 4, 6, 7). The derivatives \(\partial \overline{u}/\partial \epsilon\) of the order parameter with respect to the strain were computed with finite differences, performing a fixed-cell optimisation for each strained configuration with cell length a(ϵ) = a0(1 ± ϵ) and ϵ = 0.01, while the effective charges were computed as finite differences of polarisation. Values of polarisation along the chain (parallel to the x-axis) were computed using the Berry-phase approach, whereas components transverse to the isolated chain were computed in real space58,59. The piezoelectric coefficients cpiezo and dpiezo of Figs. 2g, h, 3, as well as the values of \({c}_{{{{\rm{piezo}}}}}^{{{{\rm{c.i.}}}}}\) and of \({c}_{{{{\rm{piezo}}}}}^{{{{\rm{i.r.}}}}}\), were computed using the Berry-phase approach60 as implemented in the code61,62, which accounts also for transverse displacements. The converse piezoelectric coefficient dpiezo is linearly related to cpiezo through the elastic constants tensor \({\mathbb{C}}\), namely \({c}_{{{{\rm{piezo}}}}}={d}_{{{{\rm{piezo}}}}}{\mathbb{C}}\). As far as 1D systems are concerned, only a single scalar elastic constant is required, and it can be evaluated as the second derivative of the energy with respect to the strain, i.e. \({\mathbb{C}}={\partial }^{2}E/\partial {\epsilon }^{2}\).
Data availability
The original data for each figure are available from the corresponding author upon reasonable request.
Code availability
All numerical calculations were performed using the CRYSTAL code. Detailed information related to the license and user guide are available here https://www.crystal.unito.it/.
References
Tadigadapa, S. & Mateti, K. Piezoelectric mems sensors: state-of-the-art and perspectives. Meas. Sci. Technol. 20, 092001 (2009).
Briscoe, J. & Dunn, S. Piezoelectric nanogenerators–a review of nanostructured piezoelectric energy harvesters. Nano Energy 14, 15–29 (2015).
Berlincourt, D. In Ultrasonic Transducer Materials (ed. Mattiat, O. E.) Ch. 2 (Springer, 1971).
Jaffe, H. Piezoelectric ceramics. J. Am. Ceram. Soc. 41, 494–498 (1958).
Rödel, J. et al. Perspective on the development of lead-free piezoceramics. J. Am. Ceram. Soc. 92, 1153–1177 (2009).
Saito, Y. et al. Lead-free piezoceramics. Nature 432, 84–87 (2009).
Setter, N. et al. Ferroelectric thin films: review of materials, properties, and applications. J. Appl. Phys. 100, 051606 (2006).
Lovinger, A. J. Ferroelectric polymers. Science 220, 1115–1121 (1983).
Ramadan, K. S., Sameoto, D. & Evoy, S. A review of piezoelectric polymers as functional materials for electromechanical transducers. Smart Mater. Struct. 23, 033001 (2014).
Vijayakanth, T., Liptrot, D. J., Gazit, E., Boomishankar, R. & Bowen, C. R. Recent advances in organic and organic-inorganic hybrid materials for piezoelectric mechanical energy harvesting. Adv. Funct. Mater. 32, 2109492 (2022).
Saxena, P. & Shukla, P. A comprehensive review on fundamental properties and applications of poly(vinylidene fluoride) (PVDF). Adv. Compos. Hybrid. Mater. 4, 8–26 (2020).
Fu, H. & Cohen, R. Polarization rotation mechanism for ultrahigh electromechanical response in single-crystal piezoelectrics. Nature 403, 281 (2000).
Kutnjak, Z., Petzelt, J. & Blinc, R. The giant electromechanical response in ferroelectric relaxors as a critical phenomenon. Nature 441, 956 (2006).
Ahart, M. et al. Origin of morphotropic phase boundaries in ferroelectrics. Nature 451, 545–548 (2008).
Damjanovic, D. A morphotropic phase boundary system based on polarization rotation and polarization extension. Appl. Phys. Lett. 97, 062906 (2010).
Liu, Y. et al. Ferroelectric polymers exhibiting behaviour reminiscent of a morphotropic phase boundary. Nature 562, 96 (2018).
Park, J. et al. Ferroelectric polymer nanofibers reminiscent of morphotropic phase boundary behavior for improved piezoelectric energy harvesting. Small 18, 2104472 (2022).
Barford, W. Electronic and Optical Properties of Conjugated Polymers (Oxford Univ. Press, 2013).
Bronstein, H., Nielsen, C. B., Schroeder, B. C. & McCulloch, I. The role of chemical design in the performance of organic semiconductors. Nat. Rev. Chem. 4, 66–77 (2020).
Onoda, S., Murakami, S. & Nagaosa, N. Topological nature of polarization and charge pumping in ferroelectrics. Phys. Rev. Lett. 93, 167602 (2004).
Kirova, N. & Brazovskii, S. Electronic ferroelectricity in carbon based materials. Synth. Met. 216, 11–22 (2016).
Rice, M. J. & Mele, E. J. Elementary excitations of a linearly conjugated diatomic polymer. Phys. Rev. Lett. 49, 1455 (1982).
Yamauchi, K. & Barone, P. Electronic ferroelectricity induced by charge and orbital orderings. J. Condens. Matter Phys. 26, 103201 (2014).
Vanderbilt, D. & King-Smith, R. D. Electric polarization as a bulk quantity and its relation to surface charge. Phys. Rev. B 48, 4442 (1993).
Xiao, D. & Niu, Q. Berry phase effects on electronic properties. Rev. Mod. Phys. 82, 1959 (2010).
Su, W. P., Schrieffer, J. R. & Heeger, A. J. Solitons in polyacetylene. Phys. Rev. Lett. 42, 1698 (1979).
Masuda, T. Substituted polyacetylenes. J. Polym. Sci., Part A: Polym. Chem. 45, 165–180 (2007).
Thouless, D. J. Quantization of particle transport. Phys. Rev. B 27, 6083 (1983).
Nakajima, S. et al. Topological thouless pumping of ultracold fermions. Nat. Phys. 12, 296–300 (2016).
Rostami, H., Guinea, F., Polini, M. & Roldán, R. Piezoelectricity and valley chern number in inhomogeneous hexagonal 2d crystals. npj 2D Mater. Appl. 2, 15 (2018).
Bistoni, O., Barone, P., Cappelluti, E., Benfatto, L. & Mauri, F. Giant effective charges and piezoelectricity in gapped graphene. 2D Mater. 6, 045015 (2019).
Romanin, D. et al. Dominant role of quantum anharmonicity in the stability and optical properties of infinite linear acetylenic carbon chains. J. Phys. Chem. Lett. 12, 10339–10345 (2021).
Ghosez, P., Michenaud, J.-P. & Gonze, X. Dynamical atomic charges: the case of ABo3 compounds. Phys. Rev. B 58, 6224–6240 (1998).
Li, Z., Antonius, G., Wu, M., da Jornada, F. H. & Louie, S. G. Electron-phonon coupling from Ab initio linear-response theory within the GW method: correlation-enhanced interactions and superconductivity in Ba1−xKxBiO3. Phys. Rev. Lett. 122, 186402 (2019).
Basko, D. M. & Aleiner, I. L. Interplay of Coulomb and electron-phonon interactions in graphene. Phys. Rev. B 77, 041409 (2008).
Lazzeri, M., Attaccalite, C., Wirtz, L. & Mauri, F. Impact of the electron-electron correlation on phonon dispersion: failure of LDA and GGA DFT functionals in graphene and graphite. Phys. Rev. B 78, 081406 (2008).
Venanzi, T. et al. Probing enhanced electron-phonon coupling in graphene by infrared resonance raman spectroscopy. Phys. Rev. Lett. 130, 256901 (2023).
Pamuk, Betül, Baima, J., Dovesi, R., Calandra, M. & Mauri, F. Spin susceptibility and electron-phonon coupling of two-dimensional materials by range-separated hybrid density functionals: case study of LixZrNCl. Phys. Rev. B 94, 035101 (2016).
Janssen, J. L., Côté, M., Louie, S. G. & Cohen, M. L. Electron-phonon coupling in C60 using hybrid functionals. Phys. Rev. B 81, 073106 (2010).
Jacquemin, D., Perpète, EricA., Ciofini, I. & Adamo, C. Assessment of recently developed density functional approaches for the evaluation of the bond length alternation in polyacetylene. Chem. Phys. Lett. 405, 376–381 (2005).
Ferretti, A. et al. Ab initio complex band structure of conjugated polymers: effects of hydrid density functional theory and GW schemes. Phys. Rev. B 85, 235105 (2012).
Barborini, M., Calandra, M., Mauri, F., Wirtz, L. & Cudazzo, P. Excitonic-insulator instability and peierls distortion in one-dimensional semimetals. Phys. Rev. B 105, 075122 (2022).
Attaccalite, C., Wirtz, L., Lazzeri, M., Mauri, F. & Rubio, A. Doped graphene as tunable electron-phonon coupling material. Nano Lett. 10, 1172–1176 (2010).
Brawand, N. P., Vörös, M., Govoni, M. & Galli, G. Generalization of dielectric-dependent hybrid functionals to finite systems. Phys. Rev. X 6, 041002 (2016).
Kronik, L. & Kümmel, S. Dielectric screening meets optimally tuned density functionals. Adv. Mater. 30, 1706560 (2018).
Refaely-Abramson, S. et al. Gap renormalization of molecular crystals from density-functional theory. Phys. Rev. B 88, 081204 (2013).
Lüftner, D. et al. Experimental and theoretical electronic structure of quinacridone. Phys. Rev. B 90, 075204 (2014).
Manna, A. K. et al. Quantitative prediction of optical absorption in molecular solids from an optimally tuned screened range-separated hybrid functional. J. Chem. Theory Comput. 14, 2919–2929 (2018).
Nakhmanson, S. M., Nardelli, M. B. & Bernholc, J. Ab initio studies of polarization and piezoelectricity in vinylidene fluoride and bn-based polymers. Phys. Rev. Lett. 92, 115504 (2004).
Ramer, N. J. & Stiso, K. A. Structure and born effective charge determination for planar-zigzag β-poly(vinylidene fluoride) using density-functional theory. Polymer 46, 10431–10436 (2005).
King-Smith, R. D. & Vanderbilt, D. Theory of polarization of crystalline solids. Phys. Rev. B 47, 1651 (1993).
Resta, R. Manifestations of berry’s phase in molecules and condensed matter. J. Condens. Matter Phys. 12, R107 (2000).
Perdew, J. P., Ernzerhof, M. & Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 105, 9982–9985 (1996).
Dovesi, R. et al. Crystal14: a program for the ab initio investigation of crystalline solids. Int. J. Quantum Chem. 114, 1287–1317 (2014).
Dovesi, R. et al. Quantum-mechanical condensed matter simulations with crystal. Wiley Interdiscip. Rev. Comput. Mol. Sci. 8, e1360 (2018).
Oliveira, D. V., Laun, J., Peintinger, M. F. & Bredow, T. Bsse-correction scheme for consistent gaussian basis sets of double-and triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 40, 2364–2376 (2019).
Weintraub, E., Henderson, T. M. & Scuseria, G. E. Long-range-corrected hybrids based on a new model exchange hole. J. Chem. Theory Comput. 5, 754–762 (2009).
Zicovich-Wilson, C. M. et al. Calculation of the vibration frequencies of α-quartz: the effect of Hamiltonian and basis set. J. Comput. Chem. 25, 1873–1881 (2004).
Pascale, F. et al. The calculation of the vibrational frequencies of crystalline compounds and its implementation in the CRYSTAL code. J. Comput. Chem. 25, 888–897 (2004).
Vanderbilt, D. Berry-phase theory of proper piezoelectric response. J. Comput. Chem. 61, 147–151 (2000).
Erba, A., El-Kelany, K. E., Ferrero, M., Baraille, I. & Rérat, M. Piezoelectricity of srtio 3: an ab initio description. Phys. Rev. B 88, 035102 (2013).
Erba, A. The internal-strain tensor of crystals for nuclear-relaxed elastic and piezoelectric constants: on the full exploitation of its symmetry features. Phys. Chem. Chem. Phys. 18, 13984–13992 (2016).
Acknowledgements
The authors acknowledge financial support from the European Union under ERC-SYN MORE-TEM, No. 951215, and from the Italian MIUR through the PRIN-2017 project, Grant No. 2017Z8TS5B. We also acknowledge CINECA awards under ISCRA initiative Grant No. HP10CCJFWR and HP10C7XPLJ for the availability of high-performance computing resources and support. Views and opinions expressed are, however, those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them.
Author information
Authors and Affiliations
Contributions
S.P.V.: theory development and implementation, numerical calculations and analysis, original draft writing. M.C.: numerical calculations and validation. P.B.: conceptualisation, validation, supervision, original draft writing. F.M.: methodology, conceptualisation, validation, supervision, and project co-ordination. All co-authors contributed to the review and editing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Villani, S.P., Campetella, M., Barone, P. et al. Giant piezoelectricity driven by Thouless pump in conjugated polymers. npj Comput Mater 10, 81 (2024). https://doi.org/10.1038/s41524-024-01269-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-024-01269-8
