
Abstract
Complete cardiac regeneration remains an elusive therapeutic goal. Although much attention has been focused on cardiomyocyte proliferation, especially in neonatal mammals, recent investigations have unearthed mechanisms by which non-cardiomyocytes, such as endothelial cells, fibroblasts, macrophages, and other immune cells, play critical roles in modulating the regenerative capacity of the injured heart. The degree to which each of these cell types influence cardiac regeneration, however, remains incompletely understood. This review highlights the roles of these non-cardiomyocytes and their respective contributions to cardiac regeneration, with emphasis on natural heart regeneration after cardiac injury during the neonatal period.
Similar content being viewed by others


Introduction
Ischemic heart disease (IHD) is the leading cause of death globally1,2. Despite the continued optimization of medical therapies and coronary revascularization strategies, the public health burden of IHD continues to grow at an alarming rate3,4,5. Cardiac regeneration has long been sought as a potential adjunctive therapy for patients with IHD. However, while various strategies to stimulate cardiac regeneration have shown promise in animal models, initial clinical trials in human patients have demonstrated inconsistent results6,7,8,9,10,11. This may be due in part to the inherent challenges of basic science research in cardiac regeneration, limitations salient to any review of evidence in this field. These include but are not limited to the difficulty of invasive procedures on neonatal rodents that are physiologically fragile and still fully dependent on their mothers post-operatively until well after the regenerative window has closed, variations in technical execution of cardiac injury (such as discrepancies in the infarct size generated, the amount of tissue resected in apical resection models, or the level of LAD ligation), significant differences in cardiomyocyte biology between the various mammalian mouse model species and humans (especially with regard to multinucleation and ploidy), a limited number of non-invasive assays, experiments, and imaging techniques, and the challenge obtaining healthy or ischemic neonatal human cardiac tissue to assess whether pathways with therapeutic promise identified in other mammals are present and function analogously in humans.
Although much research on cardiac regeneration has focused on the importance of cardiomyocyte renewal, cardiomyocytes comprise only a minority of the heart’s cells by number12,13,14. Cardiomyocyte renewal is one of the final steps in a coordinated and complex series of events involving many other cell types. Indeed, recent investigations have unearthed mechanisms by which non-cardiomyocytes, such as endothelial cells, fibroblasts, macrophages, and other immune cells, play critical roles in modulating the regenerative capacity of the injured heart. For example, in order for cardiomyocytes to repopulate the infarcted myocardium, first perfusion must be restored and cellular debris must be cleared for extracellular matrix (ECM) remodeling. The interdependency and spatiotemporal coordination of these processes during natural cardiomyocyte regeneration, as well as the degree to which each of these cell types regulates this process, remains incompletely understood. This review highlights the roles of these non-cardiomyocytes and their respective contributions to cardiac regeneration, with emphasis on natural heart regeneration after cardiac injury during the neonatal period.
Mammalian response to cardiac injury
In adult mammals, ischemic injury to the myocardium is marked by widespread cell death15. The cellular response to ischemia follow a characteristic triphasic pattern12,16. The first phase involves influx of Ly-6C(hi) macrophages and neutrophils to generate a transient high-inflammatory state characterized by elevations in IL-1β, IL-6, and TNF-α17. This is followed by a second phase in which fibroblasts and resident endothelial cells are recruited via chemokine-secreting Ly-6C(lo) macrophages12,18. Transforming growth factor-β (TGF-β) is notable among these released factors for its ability to convert fibroblasts to myofibroblasts which induce ECM production and collagen deposition, constituting early scar formation12,19,20. In the third and final phase, the recruited macrophages undergo apoptosis, collagen fibers cross-link, and the mature scar is formed12,21,22. Often there is some temporal overlap between the three mechanistically unique phases16. Unfortunately, cardiomyocyte turnover in adult mammals proceeds extremely slowly, renewing at a rate of <1% annually for most of adult life, preventing meaningful natural cardiomyocyte regeneration after injury23. Although much has been done to elucidate mechanisms of cardiac regeneration in fish and amphibians, this review focuses on what is known in mammals.
Interestingly, neonatal mammals are transiently capable of natural heart regeneration following cardiac injury. In this review, natural heart regeneration refers to the heart’s intrinsic physiologic mechanism, naturally activated in the setting of injury, to regenerate cardiac structure and function in the absence of administering exogenous factors. Neonatal mice which undergo apical resection or ligation of the left coronary artery on postnatal day 1 (P1) exhibit complete cardiac regeneration with proliferation of cardiomyocytes and minimal fibrosis, although this ability is lost if injury occurs after postnatal day 7 (P7)15,24. An apparently conserved neonatal cardiac regeneration response has also been identified in other mammalian species as well, including rats25, rabbits26, and pigs27,28. In humans, cardiomyocyte turnover and renewal has been detected in the postnatal human heart23,29,30. One study has even shown functional restoration post-injury in a neonatal human heart31. In addition to preserving normal cardiac structure and function, natural heart regeneration in neonatal mammals has also been demonstrated to restore healthy epicardial conduction dynamics and preserve native left ventricular tissue biomechanics32,33,34.
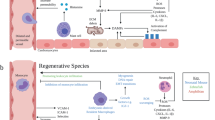
Although cardiomyocyte proliferation plays a central role in the natural regeneration response and cardiomyocytes make up 70-85% of cardiac cells by volume, they comprise only 30-40% of cardiac cells by number12. The remaining 60–70% of cardiac cells, including endothelial cells, macrophages and other immune cells, and fibroblasts, are essential contributors to clearing debris, restoring structural integrity, and facilitating reperfusion to permit proliferation of new cardiomyocytes in natural regeneration (Fig. 1). The rest of this review will focus on these three cell types.

Non-cardiomyocyte modulators of natural cardiac regeneration. Created with BioRender.com.
Endothelial cells and pericytes
Endothelial cells make up the most abundant cardiac cell type by number, accounting for over 50% of cells in the mouse and human heart, and have been highly implicated in the cardiac regeneration response35,36,37. Following injury, revascularization of the injured myocardium via migration and proliferation of vascular endothelial cells from the surrounding tissue precedes cardiomyocyte regeneration36,37,38.
Endothelial cells have been implicated and targeted in studies of the transcriptional response that occurs following infarction in adult and neonatal mice. Quaife-Ryan et al. observed that vascular endothelial cells activated a unique and robust transcriptional response after infarction in adult mice that contrasted with the neonatal response to infarction37. In the 1 day-old neonate, the transcriptional activity of three specific cell groups (vascular endothelial cells, CD90+ fibroblasts, and cardiomyocytes) associated with Wnt signaling were highly upregulated compared to their 56 day-old adult counterparts37. Interestingly, Quaife-Ryan et al. noted that Wnt-associated genes were among three of the highest-ranked neonatal transcriptional networks by expression, the other two being E2f1 and Foxm1, which are involved in cell cycle progression and proliferation37. Furthermore, they found that neonatal vascular endothelial cells were enriched with cell cycle-associated transcription factors (an upregulation of pro-mitotic transcription factors), in contrast to their adult counterparts which were enriched with C3-complement-induced transcriptional networks (an upregulation of inflammatory factors)37.
Proangiogenic effects of endothelial cells also play a role in the mammalian response to cardiac injury. Stromal cell-derived factor 1 alpha (SDF-1α) is a potent endothelial cell progenitor stem cell chemokine that initiates neovascularization after injury39. With SDF-1α as a promising target, multiple groups have investigated this chemokine as a method of exogenously-induced angiogenesis in addition to restoring biomechanical properties. In 2011, Hiesinger et al. developed an engineered SDF-1α analog (known as ESA) which was shown to effectively stimulate microrevascularization in a mouse model of cardiac injury39. Angiogenesis after myocardial injury has been associated with improved biomechanics following infarct in male Wistar rats as shown by Wang et al. in 201940. In their study, intramyocardial injection of ESA reduced infarct size, improved ventricular remodeling and function, and conserved native left ventricular biaxial mechanics40. Under hypoxic conditions, SDF-1α and other proangiogenic factors are upregulated including vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), and IL-6, improving neovascularization and functional recovery and reducing infarct size41,42,43,44,45. SDF-1α has been shown to induce chemotaxis of endothelial progenitor stem cells to stimulate neovascularization and preserve cardiac functional capacity in ovine models of myocardial infarction (MI)46. These new capillaries resulted in significantly increased arteriolar perfusion, permitting ECM retention, reducing infarct expansion, and improving left ventricular function46. Following apical resection, endothelial cells have also been shown to populate the site of injury early after resection where they generate arteries preceding cardiomyocyte growth47. Ingason et al.’s findings suggested that after injury, arterial endothelial cell-derived SDF-1α may promote a sequence of cardiomyocyte migration following angiogenesis and arteriogenesis47. In 2018, Goldstone et al. observed that while SDF-1α exerted a proangiogenic effect after infarction, it did not change the proportion of circulating bone marrow-derived endothelial cells, suggesting that SDF-1α’s proangiogenic effect likely arises via formation of new blood vessels from pre-existing endothelial cells as opposed to bone marrow-associated vasculogenesis (de novo blood vessel formation)48.
In 2019, Das et al. identified a mechanism by which neonatal mouse hearts induced collateral artery reassembly following infarct49. They found that while it was indeed associated with endothelial cells it was distinct from previously established mechanisms of de novo arteriogenesis49. The previously described mechanisms of collateral artery formation were arterialization (in which capillaries convert to arteries) and arteriogenesis (in which existing collateral arteries are widened)49,50,51. However, in this novel mechanism (termed artery reassembly) single arterial endothelial cells travel and coalesce together to generate collateral arteries following coronary ligation49. In artery reassembly, these neonatal collateral arteries form from an arterial source and in places were collateral arteries were previously absent, distinguishing this pathway from arterialization and arteriogenesis, respectively49. In 2023, Das’ group identified that a key driver of the proliferation step of artery reassembly in neonatal mouse hearts post-injury is arterial VegfR252. Their experiments demonstrated that arterial VegfR2 was downregulated in adult mouse hearts (a population in which artery reassembly does not occur after injury), and that collateral artery formation was rarely observed in arterial VegfR2-knockout models52.
While studies of revascularization after infarction have traditionally investigated pathways involving endothelial cells, the role of perivascular cells, called pericytes, remain enigmatic, in part due to the absence of unequivocal cell markers and tools for characterization and lineage tracing53. In healthy, homeostatic microvessels, pericytes and endothelial cells are separated by the vascular basement membrane and interact via peg-socket junctional complexes at fenestrations in the basement membrane to regulate the contractility of normal blood vessels54,55,56. Quijada et al. found in 2023 using multiple lineage-tracing mouse models that following myocardial infarction, pericytes traveled to the site of injury and expressed profibrotic genes55. When the same group genetically ablated Cspg4-expressing cells, a receptor downstream of the cardiac pericyte pathway they identified via single-cell RNA sequencing, they observed decreased cardiac function and increased mortality in the 2nd week following infarct55. Although comparably fewer studies have interrogated the downstream effects of pericyte activation, hyper-activation, under-activation, and deletion in the mammalian heart, early efforts have revealed promising mechanistic links between cardiac pericytes and the complex myocardial response to ischemic injury.
Macrophages and immune cells
Macrophages are mononuclear phagocytes that act as part of the innate immune system to modulate inflammation via paracrine signaling. Following tissue damage, myocardial inflammation is initiated in part by the entry and activation of macrophages, monocytes, neutrophils, T cells, B cells, natural killer (NK) cells, and other immune cells into affected myocardium57. The macrophages then experience functional and morphological changes in their roles as mediators of tissue fibrosis and repair57. Macrophages attenuate myocardial injury through scavenging of cellular debris and through secretion of cytoprotective factors such as IL-10, myeloid-derived growth factor, and fibroblast growth factor-1, which act to inhibit proinflammatory cytokines and to reduce the fibrosis response12,58,59,60,61,62. Recent studies have suggested that waves of phenotypically distinct macrophages when temporospatially coordinated play a much greater and more nuanced role than previously appreciated in natural cardiac regeneration.
In 2007, Pittet’s group observed that two functionally distinct macrophage subgroups are recruited post-ischemic injury in mouse myocardium63. Functionally, the CCR2+ macrophage response is implicated in the digestion of damaged tissue and the CX3CR1+ macrophage response facilitates tissue healing via angiogenesis and collagen deposition63. In the early phase (phase I) following infarction, Ly-6C(hi) macrophages (which are most abundant in the early phase and have pro-inflammatory function) accumulated in wild-type and CX3CR1-/- mice but were nearly absent in CCR2-/- mice, suggesting early Ly-6C(hi) macrophage accumulation in phase I relies on CCR2 (a receptor for monocyte chemoattractant protein 1, or MCP-1) but does not depend on CX3CR1 (a receptor for fractalkine)63. Ly6C(lo) macrophages, which are most abundant in the later phase and are primarily angiogenic via expression of VEGF, were found in only small quantities across all three mouse lines throughout phase I63. In contrast, during the late phase (phase II) following infarction, Ly-6C(hi) macrophages were found in low quantities across wild-type, CCR2-/-, and CX3CR1-/- mice63. However, in phase II Ly-6C(lo) accumulated in wild-type and CCR2-/- mice but failed to accumulate efficiently in CX3CR1-/- mice, indicating that in phase II, Ly-6C(lo) macrophage accumulation relies on CX3CR163.
Many studies elaborating on these findings have confirmed the importance of CX3CR1+ and CCR2+ macrophages in the healing response. A recent study by Vagnozzi et al. implicated these two macrophage subpopulations in improved heart function after ischemic injury without a corresponding increase in new cardiomyocyte production64. They found that an acute sterile immune response defined by temporal and regional induction of CCR2+ and CX3CR1+ macrophages within 8 weeks after injury improved heart function64. They also observed post-infarct mortality was significantly increased in CX3CR1-null mice, supporting the hypothesis that CX3CR1+ cells play a role in long-term pathophysiological remodeling via possible mechanisms such as influencing infarct maturation or the subsequent fibrotic response64,65. They conclude that the observed benefit to the infarcted region is based in the acute inflammation that occurs during the wound healing response, a mechanism characterized by temporary stimulation of the intrinsic wound healing cascade and macrophage subtypes64. Vagnozzi et al.’s findings support another recent paper from Epelman’s group which showed that inducible depletion of CX3CR1+ macrophages promoted pathological remodeling and increased mortality in mice, further supporting the role of these immune cells in restoration of cardiac function post-injury65. The Lavine group also recently found that tissue-resident CCR2+ macrophages promote monocyte recruitment through release of monocyte chemoattractant proteins (MCPs) with a myeloid differentiation primary response 88 (MYD88)-based mechanism as intermediary, whereas CCR2- macrophages act in opposition to inhibit recruitment of monocytes65. This is additive to previous studies which implicated direct CCR2 functional pathway inhibition in attenuation of left ventricular remodeling post-infarct66.
Although macrophages and their subpopulations (including the previously mentioned CCR2+ and CX3CR1+ populations) are essential to the regenerative response, Aurora et al. demonstrated in the neonatal mouse heart that while macrophages are not directly required for cardiomyocyte proliferation, total regeneration cannot occur without neovascularization which is dependent on macrophages, suggesting that cardiomyocyte proliferation is only one component of the functional regenerative response67. When they removed approximately half of the mononuclear phagocyte (CD11b+Ly-6G–) population in P1 neonates via clodronate liposome-mediated depletion, neovascularization deficiencies were incurred, implicating P1 macrophages in the neovascularization response post-injury67. In 2022, the Chung group showed that the macrophage subset Trem2(hi) is upregulated in the late stage after MI in adult mice, and that injection of soluble Trem2 resulted in improved cardiac function post-infarct in a similar mouse model68. In concert, these studies imply that macrophages and their subpopulations play essential roles in the regeneration process after injury, and their manipulation may provide experimental means by which therapies which strive to induce regeneration may be refined.
Other immune cell mechanisms may influence the degree of natural heart regeneration as well. A recent study found that T cell development (or transfer of adult IFN-γ-producing T-cells) impaired cardiac regeneration in neonates and was associated with greater functional and structural cardiac damage, revealing a “trade-off” relationship between cardiac regenerative potential and T cell development in neonatal mice69. T lymphocytes are largely made up of CD4+ and CD8 + T cells70. Ordinarily, CD4 + T cells activate the innate immune system, cytotoxic T cells, B-lymphocytes, non-immune cells, and include many subset cell populations with distinct roles in the natural immune response70. In non-regenerative P8 mice, ablation of CD4 + T cells was shown to increase regeneration after infarct, and neonatal mice reconstituted with adult T cells were shown to have a reduced regenerative response to alterations in the adaptive immune system71,72,73.
Regulatory T cells (Tregs), a subset of T cells that prohibit autoimmunity and maintain peripheral tolerance, have also been shown to promote cardiomyocyte proliferation in a paracrine manner74,75,76. Increasing Tregs via CD28 superagonist administration or via adoptive transfer reduced infarction-associated ventricular remodeling in mice and rats74,77,78. Specifically, Li et al. showed that the potentiation of neonatal cardiomyocyte proliferation is mediated by factors secreted from Tregs, specifically CCL24, GAS6, and AREG74. In addition, myocardial Tregs express Sparc (secreted acidic cysteine-rich glycoprotein, also called osteonectin, a matricellular protein that binds collagen), which has been shown in mouse models to increase collagen and boost infarct zone maturation resulting in a net cardioprotective effect following infarction79. The Treg results, in concert with the inverse relationship between immune maturity and regeneration, have several intriguing implications that warrant investigation. These include the potential role of immunosuppression or rheumatologic therapy in cardiac regenerative therapies, incorporating organisms with varying degrees of immune competence into experiments, and reproducing previous studies to interrogate whether better outcomes correlate inversely with immune function or activation.
Neutrophils also play a key role in the modulation of cardiac regeneration. The reparative role of neutrophils (which migrate in large proportions to the infarct site in the first few hours of ischemia) is a new and much-debated phenomenon, given that they have traditionally been associated only with pro-inflammatory damage to myocardial tissue80. The Steffens group found that following infarction, neutrophil-depleted mice had higher levels of fibrosis, decreased cardiac function, and higher likelihood of developing progressive heart failure81. They identified that a key mediator of this process was neutrophil gelatinase-associated lipocalin (NGAL) which induces macrophages with a high capacity for scavenging apoptotic cells81. When the same group restored NGAL via injection in neutrophil-depleted mice, the macrophage phenotype that promoted recovery was restored, suggesting that following infarction neutrophils polarize macrophages toward a phenotype with high ability to remove apoptotic cells, thereby improving the healing response81. Inadequate removal of apoptotic cells following MI results in adverse remodeling, delays inflammation resolution, and reduces cardiac function81,82. In addition, phagocytic clearance of necrotic myocardium and cellular debris by neutrophils promotes repair80,83.
These studies suggest that the inflammatory microenvironment generated by neutrophils may play a crucial role in the recruitment of cardiac progenitor cells84. They also highlight interesting questions of future experiments incorporating neutrophil supplementation into models of cardiac regeneration to investigate their effects on regenerative outcomes. Interrogating the relationship between selective immune cell supplementation, the use of immunosuppressive therapy, and possible interactive effects between them on mammalian models of cardiac regeneration may elucidate means by which current cardiac regenerative therapies may be improved.
Fibroblasts
Fibroblasts play a key structural role in the healthy heart and in the natural response to cardiac injury by depositing and maintaining ECM85. In healthy homeostatic myocardium, cardiac fibroblasts are considered ‘quiescent’ cells without high proliferation or ECM turnover86,87, and in adult mouse hearts CD90+ fibroblasts (the key cell type involved in fibrosis, inflammation, and cell proliferation and differentiation) comprise ~30% of nonmyocytes37,88,89. The ECM is an essential part of healthy myocardium, is comprised of a complex interconnection of components, most common among them collagens I, III, and IV, serum albumins, proteoglycans, elastins, and glycosaminoglycans, and acts as a repository for anchored growth factors, chemokines, cytokines, proteases, protease inhibitors, and noncoding RNAs90.
However, in response to hypoxia or injury, cardiac fibroblasts activate to increase synthesis of ECM constituents86,91. Excessive structural remodeling post-injury via activation of cardiac fibroblasts and the accumulation of ECM is pathophysiologically linked to scar formation, myocardial stiffening and decreased cardiac contractility92,93. Modulation of these remodeling pathways may provide a means by which natural heart regeneration is at least partly facilitated, perhaps by establishing a conducive matrix for revascularization and eventual cardiomyocyte migration.
Neonatal mouse hearts retain a robust regenerative response facilitated by inflammation and cardiomyocyte proliferation without extensive fibrosis24,86,94. Transcriptomic and phenotypic analysis of fetal human cardiac fibroblasts revealed smaller size and increased cell turnover when compared to adult human fibroblasts86,95. Transcriptomic analysis in mice also showed that compared to adult mice, embryonic mouse cardiac fibroblasts displayed higher expression of fibronectin (FN), tenascin C (TNC), collagen genes, and Postn, an expression profile associated with increased cardiomyocyte proliferation86,96. This expression profile points to fibroblasts as one of several important regulators of cardiac regeneration, though the relative contributions of fibroblasts, especially their role in the tenuous balance between sufficient fibrosis to prevent tissue from becoming aneurysmal and excessive fibrosis in the post-infarct period, remains an area of active study86,96. FN, TNC, and collagens are components of the mammalian cardiac ECM97. Postn is a protein involved in wound healing and is expressed following injury to activated fibroblasts86,98. In adult mice, post-injury ablation of Postn+ activated fibroblasts reduced fibrosis, increased cardiomyocyte abundance in the scar area, and improved cardiac function86,99. Despite the apparent cardioprotective effect of ablating Postn+ activated fibroblasts, cells expressing Postn have also been shown to be essential to the cardiac healing process following infarction, highlighting the role of Postn in cardiac regeneration as an active and incompletely understood area of research100. When Shimazaki et al. generated a mouse model of Postn knockout, they observed that after myocardial infarction there was impaired collagen fibril formation resulting in cardiac rupture, a phenotype that was rescued via gene transfer of a spliced version of Postn100. Experimentally manipulating the quantity of these ECM components using gene knockout or overexpression techniques in models of cardiac regeneration in order to assess their individual contributions to cardiomyocyte proliferation can highlight factors whose use may enhance cardiac function in current regenerative therapies.
Biomechanically, the stiffness of the ECM that forms after infarct has been shown to be inversely proportional to cardiac regenerative capacity in neonatal mice, suggesting that relative ECM composition also contributes to the regeneration response86,101,102. Increased ECM stiffness has also been proposed to inhibit the cardiomyocyte cell-cycle86,103. Cross-linking of ECM components is also an important regulator of stiffness in vivo101. Cardiomyocyte proliferation in adult mammals is also markedly lower than in neonates, and the fibrotic scar that forms and matures in absence of subsequent replacement by cardiomyocytes has been shown to pathologically reduce cardiac function and compliance86. Notably, between P1 and P2 mice, Notari et al. found that stiffness of local microenvironment is a crucial regulator of regenerative capacity even within the neonatal conserved window, observing that decreasing local ECM stiffness was associated with increased regeneration after injury101. A cellular-level explanation for this difference can be found in the Tzahor group’s paper, which demonstrates that in stiffer microenvironments, proliferating cardiomyocytes beget binucleated cardiomyocytes, in contrast to normal environments where two mononucleated cells are generated via cytokinesis from each proliferating cardiomyocyte86,102.
In 2017 transcriptomic analysis also uncovered an unexpected finding regarding the role of fibroblasts in natural regeneration. Quaife-Ryan et al. found that in adult mice, CD90+ fibroblasts retained a “transcriptional plasticity” which allowed them to regress to a “neonatal-like” state after infarction, so named because the state bears closer resemblance to neonatal cells (healthy or infarcted) than healthy adult cells37. However, they observed that adult cardiomyocytes failed to revert to neonatal transcriptional networks (especially ones implicated in cell cycle regulation), contrasting with previous dogma suggesting neonatal transcriptional reversion occurred during cardiac hypertrophy or failure in adults, and they hypothesized that reversion may be “an exception rather than the rule”37. These findings are consistent with the hypothesis that fibroblast activity and ECM deposition modulate cardiac regeneration, but the exact mechanism or mechanisms by which this occurs remains a fertile opportunity for further investigation.
The role of cardiac fibroblasts is not only limited to supporting cardiomyocyte proliferation. The Molkentin group suggested that cardiac fibroblasts are modulated by macrophage subgroups to influence restoration of mechanical function, demonstrating via gene expression analysis that isolated CCR2+ macrophages increased fibroblast expression of collagen type I α 2 (Col1a2), smooth muscle α-actin (Acta2), and lysyl oxidase (Lox)64. The expression of these three genes was reduced modestly by isolated CX3CR1+ macrophages64. However, CX3CR1+ macrophages increased connective tissue growth factor (Ctgf) expression in fibroblasts, suggesting that macrophage subgroup influences infarct structure via modulation of cardiac fibroblasts64.
Future directions
Cardiomyocyte proliferation is the central process in natural cardiac regeneration. Ultimately, cardiac regeneration depends on the generation of new cardiomyocytes and non-cardiomyocytes are likely to influence regeneration via adjunctive yet critical roles. However, in the complex crucible of cardiac regeneration, the modulation of these cardiomyocytes in addition to the modulation of functional and structural endpoints consistent with restoration of cardiac function post-injury are facilitated by a host of other cell types such as endothelial cells, macrophages, fibroblasts, T cells, and more. Given the complex interconnection of cardiac cell lines in the natural regeneration response and recent findings of improved functional restoration post-infarct in animal models by means of non-cardiomyocyte modulation of the regeneration response, future therapies may integrate our understanding of these non-cardiomyocyte cell types as new avenues to maximize functional cardiac recovery following ischemic injury.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
No new data were created in the writing of this review article.
References
Virani, S. S. et al. Heart disease and stroke statistics-2021 update: a report from the american heart association. Circulation 143, e254–e743 (2021).
Heidenreich, P. A. et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American heart association. Circulation 123, 933–944 (2011).
Ahmad, M., Mehta, P., Reddivari, A. K. R. & Mungee, S. Percutaneous Coronary Intervention 1st edn (StatPearls Publishing, Treasure Island (FL), 2023).
Alexander, J. H. & Smith, P. K. Coronary-artery bypass grafting. N. Engl. J. Med. 374, 1954–1964 (2016).
Greene, S. J. et al. Medical therapy for heart failure with reduced ejection fraction. J. Am. Coll. Cardiol. 72, 351–366 (2018).
Ang, K.-L. et al. Randomized, controlled trial of intramuscular or intracoronary injection of autologous bone marrow cells into scarred myocardium during CABG versus CABG alone. Nat. Clin. Pract. Cardiovasc. Med. 5, 663–670 (2008).
Ascheim, D. D. et al. Mesenchymal precursor cells as adjunctive therapy in recipients of contemporary left ventricular assist devices. Circulation 129, 2287–2296 (2014).
Hendrikx, M. et al. Recovery of regional but not global contractile function by the direct intramyocardial autologous bone marrow transplantation: results from a randomized controlled clinical trial. Circulation 114, I101–I107 (2006).
Menasché, P. et al. The myoblast autologous grafting in ischemic cardiomyopathy (MAGIC) trial: first randomized placebo-controlled study of myoblast transplantation. Circulation 117, 1189–1200 (2008).
Pätilä, T. et al. Autologous bone marrow mononuclear cell transplantation in ischemic heart failure: a prospective, controlled, randomized, double-blind study of cell transplantation combined with coronary bypass. J. Heart Lung Transplant 33, 567–574 (2014).
Yau, T. M. et al. Intramyocardial injection of mesenchymal precursor cells and successful temporary weaning from left ventricular assist device support in patients with advanced heart failure: a randomized clinical trial. JAMA 321, 1176–1186 (2019).
de Couto, G. Macrophages in cardiac repair: environmental cues and therapeutic strategies. Exp. Mol. Med. 51, 1–10 (2019).
Laflamme, M. A. & Murry, C. E. Heart regeneration. Nature 473, 326–335 (2011).
Varga, I., Kyselovič, J., Galfiova, P. & Danisovic, L. The non-cardiomyocyte cells of the heart. their possible roles in exercise-induced cardiac regeneration and remodeling. (ed. Xiao, J.) In Exercise for Cardiovascular Disease Prevention and Treatment: From Molecular to Clinical, Part 1 117–136 (Springer, Singapore, 2017).
Haubner, B. J. et al. Complete cardiac regeneration in a mouse model of myocardial infarction. Aging 4, 966–977 (2012).
Frangogiannis, N. G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Invest. 127, 1600–1612 (2017).
Hilgendorf, I. et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 114, 1611–1622 (2014).
Frangogiannis, N. G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 11, 255–265 (2014).
Kanisicak, O. et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7, 12260 (2016).
Serini, G. et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J. Cell Biol. 142, 873–881 (1998).
Ren, G., Michael, L. H., Entman, M. L. & Frangogiannis, N. G. Morphological characteristics of the microvasculature in healing myocardial infarcts. J. Histochem. Cytochem. J. Histochem. Soc. 50, 71–79 (2002).
Takemura, G. et al. Role of apoptosis in the disappearance of infiltrated and proliferated interstitial cells after myocardial infarction. Circ. Res. 82, 1130–1138 (1998).
Bergmann, O. et al. Evidence for cardiomyocyte renewal in humans. Science 324, 98–102 (2009).
Porrello, E. R. et al. Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080 (2011).
Wang, H. et al. Natural heart regeneration in a neonatal rat myocardial infarction model. Cells 9, 229 (2020).
Wang, H. et al. A neonatal leporine model of age-dependent natural heart regeneration after myocardial infarction. J. Thorac. Cardiovasc. Surg. 164, e389–e405 (2022).
Zhao, M. et al. Apical resection prolongs the cell cycle activity and promotes myocardial regeneration after left ventricular injury in neonatal pig. Circulation 142, 913–916 (2020).
Zhu, W. et al. Regenerative potential of neonatal porcine hearts. Circulation 138, 2809–2816 (2018).
Bergmann, O. et al. Dynamics of cell generation and turnover in the human heart. Cell 161, 1566–1575 (2015).
Sadek, H. & Olson, E. N. Toward the goal of human heart regeneration. Cell Stem Cell 26, 7–16 (2020).
Haubner, B. J. et al. Functional recovery of a human neonatal heart safter svere myocardial infarction. Circ. Res. 118, 216–221 (2016).
Wang, H. et al. Multiaxial lenticular stress-strain relationship of native myocardium is preserved by infarct-induced natural heart regeneration in neonatal mice. Sci. Rep. 10, 7319 (2020).
Wang, H. et al. Electrophysiologic conservation of epicardial conduction dynamics after myocardial infarction and natural heart regeneration in newborn piglets. Front. Cardiovasc. Med. 9, 829546 (2022).
Wang, H. et al. Natural cardiac regeneration conserves native biaxial left ventricular biomechanics after myocardial infarction in neonatal rats. J. Mech. Behav. Biomed. Mater. 126, 105074 (2022).
Colliva, A., Braga, L., Giacca, M. & Zacchigna, S. Endothelial cell–cardiomyocyte crosstalk in heart development and disease. J. Physiol. 598, 2923–2939 (2020).
Pinto, A. R. et al. Revisiting cardiac cellular composition. Circ. Res. 118, 400–409 (2016).
Quaife-Ryan, G. A. et al. Multicellular transcriptional analysis of mammalian heart regeneration. Circulation 136, 1123–1139 (2017).
Talman, V. & Kivelä, R. Cardiomyocyte—endothelial cell interactions in cardiac remodeling and regeneration. Front. Cardiovasc. Med. 5, 101 (2018).
Hiesinger, W. et al. Computational protein design to reengineer stromal cell-derived factor-1α generates an effective and translatable angiogenic polypeptide analog. Circulation 124, S18–S26 (2011).
Wang, H. et al. Bioengineered analog of stromal cell-derived factor 1α preserves the biaxial mechanical properties of native myocardium after infarction. J. Mech. Behav. Biomed. Mater. 96, 165–171 (2019).
Das, R., Jahr, H., van Osch, G. J. V. M. & Farrell, E. The role of hypoxia in bone marrow–derived mesenchymal stem cells: considerations for regenerative medicine approaches. Tissue Eng. Part B Rev. 16, 159–168 (2010).
Huang, B. et al. Myocardial transfection of hypoxia-inducible factor-1α and co-transplantation of mesenchymal stem cells enhance cardiac repair in rats with experimental myocardial infarction. Stem Cell Res. Ther. 5, 22 (2014).
Kim, S. H. et al. Hypoxia-inducible vascular endothelial growth factor-engineered mesenchymal stem cells prevent myocardial ischemic injury. Mol. Ther. J. Am. Soc. Gene Ther. 19, 741–750 (2011).
Singh, A., Singh, A. & Sen, D. Mesenchymal stem cells in cardiac regeneration: a detailed progress report of the last 6 years (2010–2015). Stem Cell Res. Ther. 7, 82 (2016).
Sullivan, K. E., Quinn, K. P., Tang, K. M., Georgakoudi, I. & Black, L. D. Extracellular matrix remodeling following myocardial infarction influences the therapeutic potential of mesenchymal stem cells. Stem Cell Res. Ther. 5, 14 (2014).
MacArthur, J. W. et al. Preclinical evaluation of the engineered stem cell chemokine stromal cell-derived Factor 1–alpha analogue in a translational ovine myocardial infarction model. Circ. Res. 114, 650–659 (2014).
Ingason, A. B. et al. Angiogenesis precedes cardiomyocyte migration in regenerating mammalian hearts. J. Thorac. Cardiovasc. Surg. 155, 1118–1127.e1 (2018).
Goldstone, A. B. et al. SDF 1-alpha attenuates myocardial injury without altering the direct contribution of circulating cells. J. Cardiovasc. Transl. Res. 11, 274–284 (2018).
Das, S. et al. A unique collateral artery development program promotes neonatal heart regeneration. Cell 176, 1128–1142.e18 (2019).
Faber, J. E., Chilian, W. M., Deindl, E., van Royen, N. & Simons, M. A brief etymology of the collateral circulation. Arterioscler. Thromb. Vasc. Biol. 34, 1854–1859 (2014).
Simons, M. & Eichmann, A. Molecular controls of arterial morphogenesis. Circ. Res. 116, 1712–1724 (2015).
Arolkar, G. et al. Dedifferentiation and proliferation of artery endothelial cells drive coronary collateral development in mice. Arterioscler. Thromb. Vasc. Biol. 43, 1455–1477 (2023).
Shen, M. et al. Stepwise generation of human induced pluripotent stem cell–derived cardiac pericytes to model coronary microvascular dysfunction. Circulation 147, 515–518 (2023).
Armulik, A., Abramsson, A. & Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 97, 512–523 (2005).
Quijada, P. et al. Cardiac pericytes mediate the remodeling response to myocardial infarction. J. Clin. Invest. 133, e162188 (2023).
van Dijk, C. G. M. et al. The complex mural cell: pericyte function in health and disease. Int. J. Cardiol. 190, 75–89 (2015).
Lafuse, W. P., Wozniak, D. J. & Rajaram, M. V. S. Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells 10, 51 (2020).
Cambier, L. et al. Y RNA fragment in extracellular vesicles confers cardioprotection via modulation of IL-10 expression and secretion. EMBO Mol. Med. 9, 337–352 (2017).
Dhingra, S., Sharma, A. K., Arora, R. C., Slezak, J. & Singal, P. K. IL-10 attenuates TNF-alpha-induced NF kappaB pathway activation and cardiomyocyte apoptosis. Cardiovasc. Res. 82, 59–66 (2009).
Korf-Klingebiel, M. et al. Myeloid-derived growth factor (C19orf10) mediates cardiac repair following myocardial infarction. Nat. Med. 21, 140–149 (2015).
Palmen, M. et al. Fibroblast growth factor-1 improves cardiac functional recovery and enhances cell survival after ischemia and reperfusion: a fibroblast growth factor receptor, protein kinase C, and tyrosine kinase-dependent mechanism. J. Am. Coll. Cardiol. 44, 1113–1123 (2004).
Shinde, A. V. & Frangogiannis, N. G. Fibroblasts in myocardial infarction: a role in inflammation and repair. J. Mol. Cell. Cardiol. 70, 74–82 (2014).
Nahrendorf, M. et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037–3047 (2007).
Vagnozzi, R. J. et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 577, 405–409 (2020).
Dick, S. A. et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 20, 29–39 (2019).
Kaikita, K. et al. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am. J. Pathol. 165, 439–447 (2004).
Aurora, A. B. et al. Macrophages are required for neonatal heart regeneration. J. Clin. Invest. 124, 1382–1392 (2014).
Jung, S.-H. et al. Spatiotemporal dynamics of macrophage heterogeneity and a potential function of Trem2hi macrophages in infarcted hearts. Nat. Commun. 13, 4580 (2022).
Dolejsi, T. et al. Adult T-cells impair neonatal cardiac regeneration. Eur. Heart J. 43, 2698–2709 (2022).
Luckheeram, R. V., Zhou, R., Verma, A. D. & Xia, B. CD4+T cells: differentiation and functions. Clin. Dev. Immunol. 2012, 925135 (2012).
Dolejsi, T. et al. Adult T-cells impair neonatal cardiac regeneration. Eur. Heart J. 41, ehaa946.3638 (2020).
Li, J. et al. Specific ablation of CD4+ T-cells promotes heart regeneration in juvenile mice. Theranostics 10, 8018–8035 (2020).
Mehdipour, M., Park, S. & Huang, G. N. Unlocking cardiomyocyte renewal potential for myocardial regeneration therapy. J. Mol. Cell. Cardiol. 177, 9–20 (2023).
Li, J. et al. Regulatory T-cells regulate neonatal heart regeneration by potentiating cardiomyocyte proliferation in a paracrine manner. Theranostics 9, 4324–4341 (2019).
Zacchigna, S. et al. Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nat. Commun. 9, 2432 (2018).
Vignali, D. A. A., Collison, L. W. & Workman, C. J. How regulatory T cells work. Nat. Rev. Immunol. 8, 523–532 (2008).
Matsumoto, K. et al. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int. Heart J. 52, 382–387 (2011).
Tang, T.-T. et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 107, 232 (2012).
Xia, N. et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation 142, 1956–1973 (2020).
Puhl, S.-L. & Steffens, S. Neutrophils in post-myocardial infarction inflammation: damage vs resolution? Front. Cardiovasc. Med. 6, 25 (2019).
Horckmans, M. et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 38, 187–197 (2017).
Wan, E. et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 113, 1004–1012 (2013).
Ma, Y. Role of neutrophils in cardiac injury and repair following myocardial infarction. Cells 10, 1676 (2021).
Cheng, B., Chen, H. C., Chou, I. W., Tang, T. W. H. & Hsieh, P. C. H. Harnessing the early post-injury inflammatory responses for cardiac regeneration. J. Biomed. Sci. 24, 7 (2017).
Tallquist, M. D. & Molkentin, J. D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 14, 484–491 (2017).
Hortells, L., Johansen, A. K. Z. & Yutzey, K. E. Cardiac fibroblasts and the extracellular matrix in regenerative and nonregenerative hearts. J. Cardiovasc. Dev. Dis. 6, 29 (2019).
Ivey, M. J. et al. Resident fibroblast expansion during cardiac growth and remodeling. J. Mol. Cell. Cardiol. 114, 161–174 (2018).
Karpus, O. N. et al. Colonic CD90+ crypt fibroblasts secrete semaphorins to support epithelial growth. Cell Rep. 26, 3698–3708.e5 (2019).
Zeng, F. et al. Role and mechanism of CD90+ fibroblasts in inflammatory diseases and malignant tumors. Mol. Med. 29, 20 (2023).
Silva, A. C., Pereira, C., Fonseca, A. C. R. G., Pinto-do-Ó, P. & Nascimento, D. S. Bearing my heart: the rule of extracellular matrix on cardiac development, homeostasis, and injury response. Front. Cell Dev. Biol. 8, 621644 (2021).
Moore-Morris, T. et al. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J. Clin. Invest. 124, 2921–2934 (2014).
Brown, R. D., Ambler, S. K., Mitchell, M. D. & Long, C. S. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu. Rev. Pharmacol. Toxicol. 45, 657–687 (2005).
Ivey, M. J. & Tallquist, M. D. Defining the cardiac fibroblast. Circ. J. 80, 2269–2276 (2016).
Porrello, E. R. et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl Acad. Sci. USA 110, 187–192 (2013).
Jonsson, M. K. B. et al. A transcriptomic and epigenomic comparison of fetal and adult human cardiac fibroblasts reveals novel key transcription factors in adult cardiac fibroblasts. JACC Basic Transl. Sci. 1, 590–602 (2016).
Ieda, M. et al. Cardiac fibroblasts regulate myocardial proliferation through β1 integrin signaling. Dev. Cell 16, 233–244 (2009).
Lockhart, M., Wirrig, E., Phelps, A. & Wessels, A. Extracellular matrix and heart development. Birt. Defects Res. A. Clin. Mol. Teratol. 91, 535–550 (2011).
Snider, P. et al. Origin of cardiac fibroblasts and the role of periostin. Circ. Res. 105, 934–947 (2009).
Kaur, H. et al. Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ. Res. 118, 1906–1917 (2016).
Shimazaki, M. et al. Periostin is essential for cardiac healingafter acute myocardial infarction. J. Exp. Med. 205, 295–303 (2008).
Notari, M. et al. The local microenvironment limits the regenerative potential of the mouse neonatal heart. Sci. Adv. 4, eaao5553 (2018).
Yahalom-Ronen, Y., Rajchman, D., Sarig, R., Geiger, B. & Tzahor, E. Reduced matrix rigidity promotes neonatal cardiomyocyte dedifferentiation, proliferation and clonal expansion. eLife 4, e07455 (2015).
Chen, W., Bian, W., Zhou, Y. & Zhang, J. Cardiac fibroblasts and myocardial regeneration. Front. Bioeng. Biotechnol. 9, 599928 (2021).
Author information
Authors and Affiliations
Contributions
B.B. wrote the first draft of the manuscript and all authors contributed significantly to revision and approval of submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baccouche, B.M., Elde, S., Wang, H. et al. Structural, angiogenic, and immune responses influencing myocardial regeneration: a glimpse into the crucible. npj Regen Med 9, 18 (2024). https://doi.org/10.1038/s41536-024-00357-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41536-024-00357-z
