
Abstract
Obesity is strongly associated with the development of diabetes mellitus and chronic kidney disease (CKD), but there is evidence for a bidirectional relationship wherein the kidney also acts as a key regulator of body weight. In this Review, we highlight the mechanisms implicated in obesity-related CKD, and outline how the kidney might modulate feeding and body weight through a growth differentiation factor 15-dependent kidney–brain axis. The favourable effects of bariatric surgery on kidney function are discussed, and medical therapies designed for the treatment of diabetes mellitus that lower body weight and preserve kidney function independent of glycaemic lowering, including sodium–glucose cotransporter 2 inhibitors, incretin-based therapies and metformin, are also reviewed. In summary, we propose that kidney function and body weight are related in a bidirectional fashion, and that this interrelationship affects human health and disease.
Key points
-
Obesity alters kidney function through multiple direct and indirect mechanisms.
-
Sodium–glucose cotransporter 2 inhibitors (SGLT2is) and glucagon-like peptide 1 receptor agonists (GLP1RAs) both promote weight loss; they also stabilize kidney function through mechanisms that are independent of their effects on weight.
-
SGLT2is induce a metabolic shift towards ketogenesis, which is likely to account for sustained weight loss.
-
The beneficial effects of GLP1RAs on the kidney are not solely explained by weight loss or improvement in glycaemic control.
-
Several GLP1R dual or polyagonist agents are under evaluation with the potential to achieve greater weight loss than GLP1RAs alone.
-
Metformin increases levels of growth differentiation factor 15 (GDF15) in the kidney and circulation to activate glial cell line-derived neurotrophic factor family receptor ɑ-like (GFRAL) in the area postrema to reduce body weight.
Similar content being viewed by others

Introduction
The global prevalence of obesity has more than doubled since 1980. In 2015, approximately 12% of the world’s adult population (641 million individuals) were classified as having obesity. If current trends continue, by 2025, approximately 18% of men and 21% of women worldwide will have obesity1. This alarming increase highlights the urgent need for effective prevention and intervention strategies to address this global public health challenge. Paralleling the increasing rates of obesity, the prevalence of chronic kidney disease (CKD) has also risen; CKD affected more than 800 million people worldwide in 2017, and remains a leading cause of morbidity and mortality2.
Obesity is characterized by the excessive accumulation of adipose tissue because of genetic, environmental and behavioural factors, and is defined as a BMI of >30 kg/m2; individuals with a BMI of >25 mg/m2 but below 30 kg/m2 are considered to have overweight. Metabolic syndrome is closely intertwined with obesity and comprises a constellation of comorbidities (Box 1), and both of these conditions predispose to cardiovascular disease (CVD)3. The term ‘cardiovascular–kidney–metabolic syndrome’ has been defined as a disorder related to the interaction of obesity, diabetes mellitus, CKD and CVD4. Importantly, the use of BMI-based definitions does have limitations, including the inability to distinguish between muscle and adipose tissue, and between different types of adipose tissue; this limitation is meaningful because visceral adiposity confers a greater risk of metabolic syndrome and CVD than does subcutaneous adiposity5.
Mendelian randomization studies have demonstrated causal associations between obesity and CKD6. Large meta-analyses have found that elevated BMI, waist circumference and waist to height ratio are independent risk factors associated with loss of kidney function (referring to the overall role of the kidney in maintaining homeostasis) and mortality in individuals with normal or reduced estimated glomerular filtration rate (eGFR)7. Even after adjustment for blood pressure and diabetes mellitus, individuals with class I obesity (BMI 30.0–34.9 kg/m2) have a 3.6-fold higher risk of end-stage kidney disease (ESKD) than those with a BMI in the normal range (18.5–24.9 kg/m2)8. Given the epidemic of obesity and CKD, interventions are urgently needed to reduce the morbidity and mortality arising from both. A number of discoveries have implicated a role for kidney-derived growth differentiation factor 15 (GDF15) in regulating weight in the hindbrain9. These findings suggest the existence of a kidney–brain–obesity axis, which has a role in modulating both feeding and body weight in a GDF15-dependent fashion.
In this Review, we begin by describing the potential mechanisms by which obesity might influence kidney function. We then outline evidence for the regulation of body weight by the kidney, and describe the effects of therapies designed for diabetes mellitus on both weight and the kidney. We thus put forward a working hypothesis that kidney function and body weight are bidirectionally and reciprocally related, which has important implications for human health and disease.
The effects of obesity on kidney function
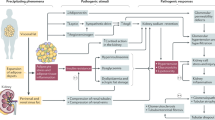
Obesity alters kidney function through direct and indirect pathways (Fig. 1). Direct effects include obesity-related glomerulopathy (ORG) and renal haemodynamic changes. Renin–angiotensin–aldosterone system (RAAS) activation, hypertension, adipokines that lead to kidney lipotoxicity, and the nucleotide-binding oligomerization-like receptor pyrin domain containing 3 (NLRP3) inflammasome might also affect kidney function.

Haemodynamic perturbations, activation of the renin–angiotensin–aldosterone system (RAAS), renal sympathetic nerve system (RSNS) activity, adipokines from visceral adipose tissue, and direct compression by renal adipose tissue all contribute to obesity-related kidney disease. RSNS activity is increased by increased leptin, decreased adiponectin and obstructive sleep apnoea leading to hypoxaemia and hypercapnia and thus downstream carotid chemoreceptor activation. Direct compression of the vasa recta and thin loop of Henle by perirenal and renal sinus adipose tissue leads to increased proximal tubular sodium reabsorption, decreased tubuloglomerular feedback and hypertension. Visceral adipose tissue produces angiotensinogen and aldosterone, resulting in the activation of RAAS. Visceral adipose tissue secretes multiple adipokines, with increases in leptin and free fatty acids and decreased adiponectin in obesity. Increased proximal tubule absorption and RAAS activation lead to decreased sodium chloride delivery to the macula densa, which reduces tubuloglomerular feedback, leading to afferent arteriolar dilation. In addition, angiotensin II (Ang-II) and aldosterone promote efferent arteriolar vasoconstriction. This combination leads to nephron glomerular hyperfiltration, with an increased glomerular filtration rate (GFR) and filtration fraction and, consequently, intraglomerular hypertension. Increased intraglomerular pressure is transmitted to podocytes, resulting in shear stress and glomerular capillary wall stress. Podocyte hypertrophy and apoptosis in response to shear stress leads to podocyte detachment and mesangial expansion, leading to the development of secondary focal segmental glomerulosclerosis termed obesity-related glomerulopathy. The combination of systemic hypertension related to RAAS activation, direct renal lipotoxic effects from adipokines from visceral adipose tissue and obesity-related glomerulopathy can lead to progressive glomerulosclerosis and tubulointerstitial fibrosis, which manifests as chronic kidney disease.
Obesity-related glomerulomegaly
In a seminal study in 1974, proteinuria was observed to remit with weight loss in four patients with obesity and nephrotic syndrome10. Focal segmental glomerulosclerosis (FSGS) is a histopathological pattern of injury characterized by segmental scarring of the glomerular capillary tuft with accumulation of extracellular matrix11. ORG refers to the pathological findings of FSGS occurring secondary to obesity, typically within hypertrophic glomeruli. Accompanying the obesity epidemic, a study in 2001 showed a tenfold rise in the incidence of ORG12. The hallmark pathological feature on renal histopathology of ORG is glomerulomegaly, which represents an adaptive response to obesity secondary to increased glomerular blood flow and hyperfiltration13,14,15. Dilation of the afferent arteriole supports the concept that adaptive hyperfiltration occurs in response to kidney injury. Altered structural and functional properties of glomeruli in ORG compromise podocyte integrity and lead to proteinuria, and nephrosclerosis occurs due to the limited ability of mature podocytes to regenerate13.
While primary idiopathic FSGS typically presents with proteinuria in the nephrotic range, lower levels of proteinuria can be observed in ORG with relatively mild foot process effacement (in which the typical interdigitating pattern of foot processes between adjacent podocytes is lost)12. This lower degree of proteinuria is probably related to the indolent development of proteinuria in secondary FSGS related to glomerular hyperfiltration in the setting of obesity16. Whereas most individuals with obesity have apparently ‘normal’ renal function according to serum levels of creatinine, in a study of kidney biopsy samples taken from individuals undergoing bariatric surgery, 38% of the patients had subclinical pathological glomerular lesions17. Moreover, despite a generally indolent clinical course, including the gradual development of proteinuria in ORG, CKD progression might still occur16.
Renal haemodynamics, RAAS activation and hypertension
Individuals with obesity exhibit altered renal haemodynamics, with afferent arteriolar vasodilation and efferent arteriolar vasoconstriction18. Consequently, these individuals have a higher GFR and filtration fraction in comparison with individuals of normal weight. Under normal physiological conditions, tubuloglomerular feedback increases afferent arteriolar vasoconstriction; however, a reduction in tubuloglomerular feedback observed in obesity leads to impairment in this autoregulation. Thus, perturbations to tubuloglomerular feedback and increased intraglomerular hypertension resulting from obesity could also predispose to the development of CKD through increases in intraglomerular pressure and shear stress18.
Systemic hypertension is common among individuals with obesity, and is partially mediated through excessive activation of the RAAS19. Adipocytes are an important component of the RAAS, as visceral adipose tissue contains angiotensinogen, the levels of which increase in parallel with increasing BMI and leptin levels; however, whether the downstream production of angiotensin II (Ang-II) is substantial enough to influence obesity-related hypertension remains uncertain20,21. In addition, the Ang-II type 1 receptor (AT1), which is responsible for efferent vasoconstriction, is present at high levels in obese rats. Thus, the combination of increased levels of ligands and receptors is likely to result in increased intraglomerular hypertension. In animal models, leptin induces aldosterone secretion, while, independent of Ang-II, adipocytes are able to stimulate aldosterone production through polyunsaturated fatty acids (such as linoleic acid) and adipokines, including C1q TNF-related protein (CTRP1)22,23. A post hoc analysis of the REIN trial found that the increased risk of ESKD in individuals with obesity compared with individuals without obesity was mitigated by treatment with the angiotensin-converting enzyme inhibitor ramipril24. Collectively, these results indicate that RAAS overactivation in obesity leads to excessive sodium reabsorption and contributes to hypertension. Thus, RAAS inhibition remains a cornerstone of treatment in obesity-associated hypertension25.
The renal sympathetic nerve system (RSNS) also contributes to hypertension in obesity, as established by the amelioration of obesity-related hypertension in response to renal denervation in dogs26. Overactivation of the RSNS has been attributed to decreased levels of adiponectin27 and increased levels of leptin28 in mice with obesity compared with those without, and to hypoxaemia and hypercapnia from obstructive sleep apnoea in humans with obesity resulting in activation of carotid body chemoreceptors and an increase in sympathetic nerve activity29.
Finally, visceral adipose tissue in the kidney, including renal sinus adipose tissue and perirenal adipose tissue deposition, might result in the direct renal compression of the vasa recta and thin loop of Henle in the medulla, leading to reduced tubular flow and increased sodium reabsorption, further pathogenically altering tubuloglomerular feedback and resulting in further hyperfiltration injury30.
Adipokines and kidney lipotoxicity
Adipocytes secrete multiple adipokines, including leptin and adiponectin, which have known paracrine and endocrine effects on kidney function; several other adipokines have been implicated in kidney function, including plasminogen activator inhibitor 1 (ref. 31) and resistin32.
Adiponectin
Adiponectin promotes fatty acid oxidation, and levels of this hormone are suppressed in states of high-fat feeding and obesity33,34. Compared with wild-type mice, adiponectin-knockout mice exhibit increased albuminuria and decreased podocyte integrity34, glomerulomegaly and tubulointerstitial fibrosis35, whereas administering adiponectin ameliorates these effects36. In podocytes, adiponectin could potentially prevent podocyte foot effacement, permeability to albumin and apoptosis by opposing NLRP3 inflammasome signalling and enhancing glomerular AMP-activated protein kinase (AMPK)-mediated regulation of oxidative stress27,34. Correspondingly, in individuals with obesity, the plasma levels of adiponectin are negatively correlated with albuminuria33.
Leptin
In contrast to the levels of adiponectin, circulating levels of leptin are increased in obesity and are associated with kidney injury, consistent with its pro-inflammatory, profibrotic and hypertensive effects37. As mentioned above, increased levels of leptin are associated with hypertension via RSNS activation28,38 and RAAS activation21,39,40. In addition, the accumulation of ectopic lipids within the kidney (owing to increased lipid storage as a consequence of excessive dietary fat intake), termed ‘renal lipotoxicity’, is implicated in ORG; renal lipotoxicity is observed as a collection of lipid droplets in podocytes, mesangial cells and proximal tubular epithelial cells41. Leptin might promote the formation of lipid droplets through upregulation of the fatty acid transporter CD36, leading to a pro-inflammatory response mediated by activation of the NLRP3 inflammasome42.
The NLRP3 inflammasome
The NLRP3 inflammasome has been implicated in the pathogenesis of both ORG and diabetic kidney disease (DKD)43,44,45,46. The NLRP3 inflammasome is a cytosolic multiprotein complex consisting of NLRP3, apoptosis-associated speck-like protein and pro-caspase 1 (ref. 45). Hyperglycaemia and hyperlipidaemia in obesity activate the inflammasome through damage-associated stimuli, including reactive oxygen species, endoplasmic reticulum stress and mitochondrial damage45,46. Upon activation, the inflammasome cleaves pro-caspase 1 into active caspase 1, which, in turn, cleaves precursors of IL-1β and IL-18 into mature inflammatory cytokines45.
In tubular epithelial cells, activation of the NLRP3 inflammasome inhibits a sirtuin 1–AMPK–lipid catabolism pathway, which increases the accumulation of lipids in the tubules and drives cellular damage47. In podocytes, NLRP3 inflammasome activation impairs autophagy and promotes the accumulation of phospholipids in dysfunctional lysosomes, leading to tubular injury44,48. Podocyte-specific NLRP3 gain-of-function diabetic mice display glomerular injury and albuminuria43, while NLRP3 deficiency or pharmacological inhibition in mice with obesity or DKD decreases the accumulation of renal lipid droplets in association with a reduction in inflammation, fibrosis and albuminuria49,50. Thus, evidence highlights the pathological role of, and the therapeutic potential of targeting, the NLRP3 inflammasome in obesity-related kidney disease.
Weight loss therapies and their effects on kidney function
Therapies designed for the treatment of diabetes mellitus, including sodium–glucose cotransporter 2 (SGLT2) inhibitors (SGLT2is) and glucagon-like peptide 1 (GLP1) receptor agonists (GLP1RAs), were initially believed to mediate weight loss and beneficial effects on the kidney and cardiovascular system through their anti-hyperglycaemic effects. However, multiple studies have shown that these favourable effects are largely independent of any glucose-lowering ability. Metformin also results in appreciable weight loss in individuals with or without diabetes mellitus, with a weight reduction of 2.1 ± 5.7% observed in the Diabetes Prevention Program51,52. Bariatric surgery remains the most effective intervention for sustained weight loss over a decade53. In the following sections, we review the mechanisms by which bariatric surgery, SGLT2is, incretin-based therapies and metformin might affect weight loss and the kidney.
Bariatric surgery
Bariatric surgery, including Roux-en-Y gastric bypass and sleeve gastrectomy, confer both enhanced quality of life and survival in individuals with obesity53. Beyond weight loss, bariatric surgery can induce remission of diabetes mellitus, normalize blood pressure and attenuate the progression of obesity-related CKD54,55,56. In the GATEWAY trial, which evaluated improvement in hypertension in 100 patients with obesity and hypertension, the combination of Roux-en-Y gastric bypass and medical therapy was more effective than medical therapy alone at improving blood pressure57. Postulated mechanisms by which bariatric surgery exerts renoprotective effects include sustained weight loss and its associated metabolic effects in addition to a reduction in blood pressure (Fig. 2). Notably, the beneficial effects of bariatric surgery on kidney function might not be entirely attributable to weight loss. A meta-analysis found that improvements in albuminuria after bariatric surgery were not correlated with changes in weight or glucose levels, suggesting that bariatric surgery can improve kidney function independent of weight loss and glycaemic control58. In addition, bariatric surgery has been associated with decreased levels of serum creatinine and normalized eGFR, and these effects were associated with improved blood pressure and metabolic parameters, such as glycaemic control, rather than reductions in weight or BMI55.

Bariatric surgery alters circulating levels of adiponectin, leptin and growth differentiation factor 15 (GDF15), and in parallel reduces perirenal and renal sinus adipose tissue. Decreased perirenal and renal sinus adipose tissue could lead to a reduction in the accumulation of renal lipids, which decreases lipotoxicity and nucleotide-binding oligomerization-like receptor pyrin domain containing 3 (NLRP3) inflammasome activation. Changes in adiponectin and leptin levels after bariatric surgery might confer renoprotective effects, as adiponectin can preserve podocyte integrity and reduced leptin levels might decrease hypertension, renal sympathetic nerve system (RSNS) activity and renin–angiotensin–aldosterone system (RAAS) activation. Increased circulating levels of GDF15 have been associated with decreased body weight after surgery. GDF15 might also act as an anti-inflammatory and antifibrotic factor in the kidney.
Bariatric surgery could exert beneficial effects on the kidney via alterations in adipokine signalling. Following sleeve gastrectomy in individuals with moderate to severe CKD, reductions in proteinuria and insulin resistance were associated with increased levels of adiponectin and decreased levels of leptin59. Beyond improving insulin resistance, adiponectin might also directly preserve podocyte integrity, given the negative correlation between adiponectin and albuminuria in obesity27,33. Decreased levels of leptin might also result in anti-hypertensive effects by reducing RSNS and RAAS activation28,39,40,60.
Consistent with the postulated direct compressive effects of increased renal adipose tissue on renal vasculature and tissue to alter tubular flow and sodium reabsorption21, the amount of renal sinus adipose tissue is negatively associated with eGFR in obesity61. Furthermore, decreases in both renal sinus adipose tissue and perirenal adipose tissue have been associated with remission of hypertension following bariatric surgery61,62. Decreased renal adipose tissue after bariatric surgery might also benefit obesity-related kidney disease through decreased lipotoxicity and decreased NLRP3 inflammasome activation; however, these potential renoprotective effects remain to be directly studied.
Although future studies are required, we propose that bariatric surgery stabilizes renal function partially by decreasing the accumulation of renal lipids and levels of leptin, as well as increasing adiponectin levels. Collectively, these mechanisms might contribute to the preserved glomerular function observed following bariatric surgery, ultimately decreasing albuminuria and the development of CKD7,63.
SGLT2is
SGLT2is were developed for the treatment of type 2 diabetes mellitus (T2DM) as a therapy for glycaemic control. SGLT2 is predominantly expressed in the S1 and S2 segments of the proximal tubule, where it functions as a sodium-dependent glucose transporter to reabsorb up to 90% of glucose from the glomerular filtrate; consequently, its inhibition results in glucosuria and calorie loss64. Although these agents were designed for the treatment of T2DM, they only modestly improve glycaemic control, lowering HbA1c by 0.5–1.0% on average65. Unexpectedly, however, considerable benefits predominantly on heart failure and kidney outcomes were consistently observed across trials in patients with different levels of cardiovascular and kidney risk66,67.
Kidney benefit
One of the main mechanisms by which SGLT2is confer benefit in the kidney involves activation of tubuloglomerular feedback, wherein decreased glucose and sodium reabsorption in the proximal tubule in response to SGLT inhibition increases the distal delivery of sodium chloride to the juxtaglomerular apparatus. Feedback to the macula densa results in afferent vasoconstriction via adenosine, reducing intraglomerular hypertension and preventing long-term glomerular injury in individuals with type 1 diabetes mellitus (T1DM) and hyperfiltration68. However, in individuals with T2DM and preserved kidney function, the RED trial found that the SGLTi dapagliflozin reduces GFR without affecting renal vascular resistance, suggesting that post-glomerular efferent arteriolar vasodilation might be the underlying haemodynamic mechanism leading to benefit in this population69. Furthermore, hyperglycaemia is associated with increased proximal tubular work and oxygen consumption through increased sodium–glucose cotransport coupled to Na+/K+-ATPase activity. In diabetic rats, treatment with the non-selective SGLTi phlorizin reduced renal blood flow, Na+/K+-ATPase activity and oxygen consumption70. The tubular hypothesis postulates that SGLT2is confer kidney benefit through reduced energy expenditure on proximal tubule transport, thereby reducing renal cortical oxygen consumption71. The striking cardiovascular benefit of SGLT2is also led to the ‘thrifty substrate’ hypothesis, which postulates that ketogenesis induced by SGLT2is alters metabolism to utilize β-hydroxybutyrate instead of fatty acids as an oxidation substrate in the heart and kidney, thereby improving oxygen consumption efficiencies and mediating cardioprotective effects72.
CREDENCE, DAPA-CKD and EMPA-Kidney were landmark trials that evaluated the efficacy of SGLT2is in terms of primary kidney end points, providing clear evidence for their benefit across the spectrum of CKD, including in individuals without diabetes mellitus66,67,73. The CREDENCE trial evaluated the SGLT2i canagliflozin in patients with T2DM with an eGFR of 30 to <90 ml/min/1.73 m2 and albuminuria (urine microalbumin to creatinine ratio (UACR) 300–5,000 mg/g) on a background of RAAS blockade, and found a reduction in the composite of doubling of serum levels of creatinine, ESKD and death from renal or cardiovascular causes by 30% compared with the placebo group67. The DAPA-CKD study expanded the study population to include adults with either DKD or non-DKD with an eGFR of 25–75 ml/min/1.73 m2 and a UACR of 200–5,000 mg/g on a background of RAAS blockade73. In this trial, dapagliflozin reduced the primary composite outcome of a sustained decline in the estimated GFR by >50%, ESKD and renal or cardiovascular death by 39% without heterogeneity by diabetes status compared with placebo. The EMPA-Kidney trial further evaluated the benefit of SGLT2is in individuals without albuminuria by including adults with or without diabetes mellitus with an eGFR of 20–45 ml/min/1.73 m2 regardless of albuminuria or an eGFR of 45–90 ml/min/1.73 m2 with a UACR of ≥200 mg/g on a background of RAAS blockade66. In this trial, 6,609 patients were enrolled and followed for a median of 2 years; a 28% lower risk of the primary composite outcome of ≥40% decline in eGFR, ESKD or death from renal or cardiovascular causes was reported with empagliflozin treatment compared with placebo, similarly without heterogeneity by diabetes status or eGFR subgroups. All-cause hospitalizations were also reduced by 14% with empagliflozin treatment; however, heart failure hospitalization or cardiovascular death did not differ significantly between the treatment group and placebo group, although this apparent lack of difference might have been affected by lower event rates owing to early trial discontinuation for efficacy.
Weight loss
The use of SGLT2is consistently brings about significant body weight loss. Glucosuria results in 1 kg of weight loss within a week of initiation of SGLT2i treatment and leads to depletion of glycogen and adipose stores from the liver and skeletal muscle. In response to SGLT2 inhibition, the pancreas increases glucagon levels, with a reduction in the glucagon to insulin ratio. In adipose tissue, lipolysis occurs, in addition to augmentation of adipose tissue browning, thereby increasing energy expenditure. Over the longer term, a total of 2–3 kg of weight loss occurs with SGLT2i treatment, with a nadir at approximately 6 months. In a substudy conducted in 660 participants in the EMPA-Kidney trial, empagliflozin reduced fluid overload but had no significant effect on adipose tissue mass, as determined by bioelectrical impedance analysis over 18 months74. Glucosuria induced by SGLT2i treatment results in the loss of 60–80 g of glucose daily, which corresponds to 1,000–1,300 kJ (230–310 kcal) per day. If sustained, this negative energy balance would be expected to result in the loss of 7 kg over a 6-month period, although this loss might be offset by a compensatory increase in the consumption of carbohydrate-rich foods75,76. In diet-induced obese rats, dapagliflozin treatment was accompanied by compensatory hyperphagia, which attenuated the magnitude of weight loss, and in adult humans, dapagliflozin treatment leads to an increase in daily sucrose intake77. In 86 human participants treated with empagliflozin, chronic glucosuria resulted in an adaptive increase in energy intake78. Thus, small increases in energy intake might fully offset energy losses related to glucosuria, whereas increased dietary adherence might further promote weight loss in the context of SGLT2 inhibition. However, this hypothesis has been disputed by the results of two further trials. The ENERGIZE trial found that a reduction in body weight and liver fat in 52 patients treated with dapagliflozin was not associated with compensatory adaptations in food intake or energy expenditure79. Furthermore, the SEESAW trial in individuals with overweight or obesity and T2DM found no alterations in subjective measures of appetite or postprandial appetite-regulatory gut peptides (total peptide YY, GLP1 and acylated ghrelin) after 24 weeks of empagliflozin treatment80. Therefore, an alternative hypothesis to explain the steady state of weight loss achieved with SGLT2is might be that weight loss itself results in metabolic adaptations that lead to lower energy expenditure to compensate for the energy lost through glucosuria81.
Aestivation, weight loss and kidney function
Although SGLT2is induce osmotic diuresis through glucosuria and accompanying sodium natriuresis, long-term diuresis with SGLT2i treatment is not observed owing to adaptive responses by the RAAS and vasopressin systems82. Osmotic diuresis is instead coupled to an aestivation-like state to maintain fluid homeostasis through an integrated response linked to long-term regulation of body weight (Fig. 3), which appears to have beneficial effects on organ preservation. Aestivation refers to a state of dormancy under arid conditions to allow energy and water conservation to sustain life. During this aestivation-like state, the metabolic rate decreases83 and organic osmolytes counteract osmotic diuresis to maintain hydration status via urea gradient-driven water reabsorption84, ureagenesis in the liver and muscle resulting in increased energy expenditure85, and alteration in cardiovascular energetics, all of which are outlined below.

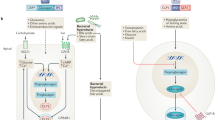
Sodium–glucose cotransporter 2 inhibitors (SGLT2is) inhibit SGLT2 in the proximal convoluted tubule to induce glucosuria, resulting in a negative energy balance and activating hypometabolic aestivation-like patterns that couple energy expenditure with preservation of fluid balance. SGLT2i use results in immediate weight loss with depletion of glycogen and adipose stores from the liver and skeletal muscle. With chronic use, ketones are prioritized as the energy substrate over glucose, a process that is coupled to the production of osmolytes to facilitate water reabsorption in the kidney. The liver prioritizes ketogenesis, resulting in ureagenesis. Skeletal muscle provides the liver with amino acids, including branched-chain amino acids (BCAA), through the glucose–alanine shuttle (the Cahill cycle) without loss of lean muscle mass. BCAAs are specifically catabolized in the heart and kidney to short-chain fatty acids and ketones as an energy source. Osmotic diuresis associated with glucosuria and sodium chloride is offset by the production of osmolytes, including urea, through ketogenesis in the liver, which facilitates water reabsorption, and thus diuresis is attenuated. Water reabsorption occurs through vasopressin-mediated mechanisms, including increased expression of the urea transporter A1 (UT-A1), which supports water conservation by driving the urea countercurrent exchange system in the renal medulla in an energy-efficient manner, and is probably accompanied by a reduction in metabolic rate in the renal cortex. In the pancreas, SGLT2is increase glucagon levels and reduce the glucagon to insulin ratio, supporting alterations in metabolism. In adipose tissue, increased browning and lipolysis increase energy expenditure. Compensatory increases in the central regulation of appetite occur. These mechanisms collectively maintain weight loss despite ongoing glucosuria.
Osmolytes retain fluid in body compartments through osmotic activity, with sodium and chloride being the most important owing to their extracellular abundance. To avoid volume depletion as a consequence of sodium-rich osmotic diuresis induced by SGLT2is, vasopressin maintains the fluid balance by mediating the active accumulation of urea (another osmolyte) in the renal medullary interstitium through the increased expression of the urea transporter A1, and activates aquaporin 2 to enhance water reabsorption86. The increased energy needed for the production and accumulation of urea osmolytes results in a reprioritization of energy expenditure, and explains why further weight loss is not observed with chronic SGLT2i use despite ongoing glucosuria86. Although impaired kidney function, such as is seen in individuals with CKD, attenuates the magnitude of glucosuria with SGLT2i treatment, weight loss is independent of this glycaemic lowering87.
The hepatic response to SGLT2i treatment includes reprioritization of ketones (produced in the liver by the breakdown of fatty acids) as an energy substrate over glucose, a process that also results in the production of urea through the urea cycle, offsetting urinary solute loss. The production of urea requires nitrogen (in addition to energy) and, in this context, skeletal muscle serves as a nitrogen source, providing the liver with amino acids, including branched chain amino acids, through the glucose–alanine shuttle (the Cahill cycle) without any loss of lean muscle mass. The branched chain amino acids are then catabolized as an energy source specifically in the heart and kidney (not in the liver, as hepatocytes lack a branched chain amino acid transferase required for catabolism) to short chain free fatty acids and ketone bodies83. Thus, SGLT2i treatment is hypothesized to mobilize branched chain amino acids from skeletal muscle specifically as a fuel source for the heart and kidney. Another consequence of this altered metabolism with SGLT2i treatment on the use of free fatty acids in preference to glucose is a reduction in lipid metabolites, thereby maintaining podocyte health by reducing insulin resistance as well as reducing oxidative stress, inflammation and fibrosis in the kidney88.
In response to SGLT2i treatment, augmentation of adipose tissue browning via the induction of polarization of M2 macrophages in the white adipose tissue and the liver is observed, resulting in increased energy expenditure89. Furthermore, autophagy, the physiological process by which organelles and proteins undergo proteolytic degradation in lysozymes, is important for cell survival. Autophagic flux is promoted by SGLT2is, and some of the benefits observed during the aestivation metabolic switch might be mediated through autophagy induced by reduced mTOR signalling. In mouse models, mTOR1C1 overactivation, which reduces autophagic flux, results in impaired renal lipolysis and downstream kidney damage, which can be abrogated by SGLT2i-mediated increases in the production of ketone bodies90. In summary, the aestivation-like state induced by SGLT2is alters metabolism towards ketogenesis to maintain long-term weight loss and is coupled to the preservation of fluid balance.
Incretin-based therapies
Incretins are a class of gut hormones that stimulate the secretion of insulin from pancreatic β-cells in a glucose-dependent manner, and include glucose-dependent insulinotropic polypeptide (GIP) and GLP1. The actions of both GIP and GLP1 have been exploited in the treatment of T2DM91. Human GLP1 undergoes rapid proteolytic degradation in the circulation by dipeptidyl peptidase 4 (DPP4), which initially limited its therapeutic application. However, exendin 4, which is derived from the saliva of the Gila monster Heloderma suspectum, is homologous to human GLP1 but is resistant to DPP4-mediated degradation. This homology and resistance to degradation have enabled the development of human GLP1 analogues modified to prolong the duration of action, and synthetic exendin 4-based GLP1RAs have thus facilitated the clinical use of GLP1RAs. In addition to conferring beneficial effects on glycaemic control, GLP1RAs have positive effects on vascular function and lipid profile as well as reducing blood pressure and appearing to attenuate the process of atherosclerosis92. Cardiovascular outcome trials have demonstrated that this class of agents reduces the risk of major adverse cardiovascular events in individuals with T2DM compared with placebo93.
GLP1RAs and weight loss
GLP1RAs are potent anti-obesity agents, but the mechanism by which they promote weight loss is independent of their effects on glycaemic control. Network meta-analyses have demonstrated that GLP1RAs, particularly semaglutide, are highly effective for weight management, with a body weight change of 5.8% from baseline over a median of 24 weeks94. Weight loss associated with GLP1RAs occurs through central effects that reduce energy intake by decreasing appetite and calorie intake, and promoting satiety95,96,97.
GLP1R signalling in feeding and the regulation of body weight is initiated in the paraventricular nucleus and the dorsal vagal complex of the brain98,99. Additional results indicate that the GLP1RA liraglutide is internalized upon binding to the GLP1R and alters hypothalamic neurons to affect energy balance100, while GLPR signalling in the nucleus of the solitary tract is necessary for liraglutide to reduce feeding and weight101. Although all GLP1RAs induce weight loss, there are considerable differences between the agents, with semaglutide inducing weight loss to the greatest extent. Structural differences might underlie differences in body weight loss potency and affect passage across the blood–brain barrier; smaller and longer-acting GLP1RAs, such as liraglutide and semaglutide, potentially have a more potent effect on the central regulation of appetite than larger, shorter-acting GLP1RAs102.
GLP1RAs and the kidney
Secondary analyses of cardiovascular outcome trials have found that GLP1RAs reduce albuminuria103. However, their effects on glycaemic control and weight loss do not fully account for the mechanisms of action of GLP1RAs in the kidney104. Evidence from animal models demonstrates that GLP1RAs can activate protein kinase A, which, in turn, inhibits NADPH oxidase and nuclear factor κB, thereby reducing oxidative stress and inflammation105. Downregulation of pro-inflammatory pathways and cytokines associated with obesity, including tumour necrosis factor-α, monocyte chemoattractant protein 1 and fibronectin, might be another putative mechanism by which GLP1RAs mediate their beneficial effects in the kidney102. In addition, the potential effect of GLP1RA treatment in improving renal hypoxia is being explored, as these agents might attenuate the rarefaction of vasculature that occurs with DKD106.
In terms of more direct effects in the kidney, GLP1RAs have been shown to inhibit the sodium–hydrogen exchanger 3 in the proximal renal tubule107. However, tubuloglomerular feedback is not a major mechanism by which GLP1RAs mediate their benefit in the kidney, as infusion with the GLP1RA exenatide does not alter renal haemodynamics in patients with T2DM despite increasing proximal sodium excretion108. The FLOW trial (NCT03819153), which aimed to evaluate the effect of semaglutide on DKD in 3,534 adults, was discontinued early for efficacy in achieving a primary composite kidney end point; the results are expected to be published in the first half of 2024 (ref. 109).
GLP1 polyagonists
The success of GLP1-based therapies has prompted the evaluation of dual agonists or polyagonists that target receptors for GIP, glucagon, amylin or peptide YY in combination with GLP1RAs (Table 1). Through actions on multiple receptors in different target tissues, a greater extent of weight loss and glucose reduction is observed in comparison with GLP1RAs alone.
Tirzepatide is a GLP1R and GIPR imbalanced dual agonist that preferentially activates the GIPR110. In the SURPASS-4 trial, tirzepatide reduced the UACR and attenuated the rate of eGFR decline meaningfully compared with insulin glargine111.
Triple agonists that target GLP1, GIP and glucagon receptors with the aim of further reducing food intake and weight loss are under evaluation112. The results of a phase II study of triple agonism of the GLP1, GIP and glucagon receptors using retatrutide (LY3437943) at multiple weekly doses found that treatment with the highest dose (12 mg) resulted in a −24.2% least-squares mean percentage change in body weight at 48 weeks (compared with −2.1% in the placebo group), supporting further potential efficacy for weight loss with the addition of glucagon receptor agonism113. The mechanism(s) responsible for the metabolic benefits of GLP1R-based polyagonists remain largely unknown, although, in addition to the weight-lowering effect of GLP1, bidirectional changes in GIP and glucagon signalling in the brain also alter feeding and weight114,115,116,117,118. Surprisingly, given the efficacy of GLP1R and GIPR dual agonists, a phase I study of AMG133, a bispecific molecule that agonizes GLP1R and antagonizes GIPR in vitro, has shown promising results in inducing weight loss119.
Endogenous amylin is co-secreted with insulin and has roles in delaying gastric emptying, inhibiting glucagon release and reducing appetite120; the combination of a long-acting amylin analogue, cagrilintide, and semaglutide resulted in greater weight loss than semaglutide alone121. Similarly, the combination of semaglutide with a peptide YY analogue (NNC0165-1875), which acts as an agonist on the NPY receptor type 2 (Y2R) to centrally regulate food intake122, has demonstrated efficacy in murine models, and has recently been investigated in humans (NCT04969939).
Mechanistic studies, such as REMODEL (NCT04865770) and TREASURE-CKD (NCT05536804), aim to provide more comprehensive information on the potential mechanism(s) of renal benefit of GLP1RAs, including blood oxygen level-dependent MRI of the kidneys, which measures renal tissue deoxyhaemoglobin levels as a functional, validated measure of renal hypoxia123. Future studies are also warranted to investigate the overlapping and non-overlapping mechanism(s) of GLP1R-based polyagonists versus GLP1RAs in regulating energy balance.
In summary, SGLT2is and GLP1RAs lower weight, have beneficial effects on the kidney and reverse obesity-induced complications. However, both therapies might confer their renal benefit independent of weight changes, which raises the question of whether the effects of these therapies in the kidneys might mediate the effects on weight loss. Next, we discuss findings indicating that metformin indeed directly acts on the kidney to influence weight loss.
Metformin
Metformin is a first-line therapy for T2DM that lowers glucose levels primarily by lowering hepatic glucose production124. As mentioned above, metformin also results in appreciable weight loss in individuals with or without diabetes mellitus. The mechanism(s) responsible for the weight-lowering effect of metformin remain controversial, although GDF15 and its hindbrain receptor, glial-derived neurotrophic factor receptor ɑ-like (GFRAL), have been implicated as potential mediators9,125,126.
GDF15 action in the brain
GDF15 is a divergent member of the transforming growth factor-β superfamily127,128 that is expressed in the liver, kidney and intestine, and mediates the integrated stress response126,129,130,131. Injection of recombinant GDF15 or GDF15 overexpression lowers food intake and body weight in lean and obese mice as well as in monkeys through the activation of its receptor, GFRAL132,133,134,135,136,137. GFRAL is almost exclusively expressed in the area postrema and nucleus of the solitary tract of the hindbrain132,133,134,135,138 and recruits the receptor tyrosine kinase, RET, as a co-receptor to mediate cellular signalling132,133,134. Activation of GFRAL in the area postrema, but not in the nucleus of the solitary tract, is sufficient to lower food intake and weight9,138.
Similar to GLP1RAs, GDF15 alters food preferences in individuals with obesity and contributes to reduced caloric intake in mice131,132,139,140. In addition to suppressing appetite, recombinant GDF15 mediates weight loss in mice through a GFRAL–β-adrenergic receptor signalling pathway that counteracts compensatory reductions in energy expenditure (in response to reduced intake) in skeletal muscle136 as well as via delayed gastric emptying137 and enhanced adipose tissue browning141.
A relationship between metformin and GDF15 was first implicated in a study of 2,317 individuals receiving metformin. Of the 237 biomarkers that were evaluated, GDF15 had the strongest association with metformin use142. Indeed, the use of metformin increases plasma levels of GDF15 in association with weight loss in individuals with overweight or obesity with or without T2DM125,126. Oral metformin in rodents increased circulating levels of GDF15 and reduced food intake, body mass, fasting insulin and glucose intolerance through the activation of GFRAL9,125,126.
To address whether GDF15–GFRAL signalling in the area postrema of the brain mediates the effect of the metformin-induced increase in GDF15, it was first confirmed that a single dose of metformin increased plasma levels of GDF15 and lowered feeding and weight in male rats fed a high-fat diet9. Abrogating the expression of GFRAL in the area postrema negated the ability of GDF15, as well as metformin, to lower food intake and weight, indicating that metformin increases circulating levels of GDF15 to activate GFRAL in the hindbrain to lower food intake and weight9.
The kidneys as a source of GDF15
Studies indicated that the release of GDF15 from the kidneys mediates the increase in plasma levels of GDF15 in response to metformin and is necessary to lower feeding and body weight in rats9. Specifically, knocking down Gdf15 expression in the kidney prevented acute or 11-day metformin administration from increasing plasma GDF15 levels and lowering food intake and weight in rats9. The cell types responsible for the production of GDF15 in the kidney are likely to be in the tubulointerstitial compartment, as human kidney biopsies revealed a greater relative abundance of Gdf15 mRNA expression in tubulointerstitial tissue than in glomerular tissue9. In addition, in situ hybridization revealed that metformin induced Gdf15 expression in the periglomerular renal tubular epithelial cells of mice126. As well as metformin, cellular stressors such as hypoxia and aberrant protein folding also induce Gdf15 expression through the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) and upregulation of the downstream transcriptional regulators ATF4 and C/EBP homologous protein (CHOP)130. Metformin might increase GDF15 levels through the eIF2α–ATF4–CHOP axis, as the expression of kidney-derived Gdf15, Chop and ATF4 protein is upregulated in metformin-treated rodents9,126; however, future studies investigating the potential mechanisms underlying the regulation of Gdf15 are warranted (Fig. 4).

Studies predominantly in rodents have demonstrated that metformin upregulates Gdf15 expression in the kidney to increase circulating levels of growth differentiation factor 15 (GDF15), which activates glial cell line-derived neurotrophic factor family receptor α-like (GFRAL) receptors in the area postrema (AP) of the hindbrain. GDF15-bound GFRAL recruits RET, a tyrosine kinase receptor, as a co-receptor to mediate central signalling that leads to decreased food intake and, thus, reduced weight gain. Phosphorylation of eukaryotic initiation factor 2α (eIF2α) induces the binding of transcription factors ATF4 and CHOP to the GDF15 gene promoter to upregulate Gdf15 expression, although whether metformin increases the expression of Gdf15 in the kidney through this mechanism is unclear. NTS, nucleus tractus solitarius.
The identification of the kidney as a key source of GDF15 in metformin treatment reveals a novel kidney–brain axis in the regulation of energy balance. Future studies are needed to assess whether these findings translate to humans. Collectively, these studies characterize a previously unrecognized role for the kidney in the regulation of food intake and weight. Together with the fact that increased weight gain disrupts kidney function as outlined above, we propose that kidney function and body weight are bidirectionally and reciprocally related.
Metformin, GDF15 and kidney function
In patients with advanced DKD, metformin reduces albuminuria and progression to ESKD, but might rarely be associated with lactic acidosis in advanced CKD; thus, the benefit of metformin in this context remains controversial143. The renoprotective effects of metformin have been attributed to reduced lipid accumulation and reduced fibrosis via the activation of AMPK signalling144. In addition, metformin-induced AMPK activation in the kidneys of diabetic mice leads to enhanced autophagy and mitophagy in association with decreased markers of tubulointerstitial fibrosis, oxidative stress and mitochondrial damage145. However, metformin maintains the ability to reduce inflammatory markers and to promote an antioxidant response in mice that are deficient in the β1 subunit of AMPK and thus showed compromised AMPK activity in the kidney146, suggesting that metformin might also act through AMPK-independent mechanisms to confer protection from kidney damage.
The ability of metformin to increase the levels of Gdf15 in the kidney might mediate some of its renoprotective benefits147,148. A potential protective mechanism for GDF15 in the kidney might involve antifibrotic actions, as overexpression of GDF15 or its systemic administration in kidney injury attenuates markers of renal inflammation, cell death and fibrosis148; GDF15 could upregulate or sustain the expression of Klotho, an anti-inflammatory and antifibrotic protein that is abundantly expressed in the kidney but downregulated in rodent models of kidney injury148.
In murine models of both T1DM and T2DM, knockout of GDF15 leads to tubulointerstitial damage and subsequent glucosuria and polyuria147. However, in a meta-analysis of 14 studies, elevated levels of GDF15 were also found to be associated with CKD progression149. Furthermore, patients with stage 3–5 CKD had circulating levels of GDF15 that were approximately twofold higher than those of control individuals150. Increases in GDF15 levels might therefore represent an initial adaptive response to acute stress, diabetes mellitus and obesity, which might become maladaptive in chronic disease states, including ESKD or malignancy, in which elevations in GDF15 contribute to anorexia–cachexia syndrome151. Increases in GDF15 in ESKD might relate to its decreased renal clearance or a compensatory increase to protect against kidney injury148,152. Conversely, in patients who had undergone kidney transplantation, a decrease in GDF15 was observed 1 year after the procedure152. Assessing whether metformin still induces weight loss in individuals with CKD would be informative, given that ESKD is associated with cachexia rather than weight gain. Future investigations are warranted to clarify the inter-relationship between metformin, GDF15, kidney function and the risk of CKD.
Bariatric surgery and GDF15
GDF15 levels are also increased in response to bariatric surgery. Roux-en-Y gastric bypass in obese diabetic rats resulted in increased systemic and portal vein levels of GDF15 28 days after surgery compared with weight-matched, food-restricted rats; this increase was inversely associated with postoperative weight and food intake153. Although the increase in portal vein GDF15 levels implies increased secretion from the small intestine, systemic GDF15 levels were higher than portal GDF15 levels by approximately threefold, indicating that other tissues were also likely to have contributed to the systemic increase in GDF15 in rats that had undergone Roux-en-Y gastric bypass153. This observation has been replicated in humans: GDF15 levels were seen to increase as soon as 1 month following sleeve gastrectomy and were negatively associated with changes in BMI154,155. In a study in 47 individuals with and without diabetes mellitus who underwent Roux-en-Y gastric bypass, circulating levels of GDF15 increased within 1 week of surgery and remained elevated after 4 years, correlating with the degree of body weight loss156. Thus, future studies investigating the increase in human and rodent GDF15 levels after bariatric surgery to assess their role in maintaining surgically-induced weight loss are warranted; further investigation of the relative contribution of GDF15 from different organs, including the kidney, is also warranted9. Experimental findings that GDF15 in the kidney decreases markers of renal inflammation, cell death and fibrosis147,148 further justifies future studies investigating the changes in kidney GDF15 expression after bariatric surgery.
GDF15 as a potential weight loss therapy?
Given the role of GDF15 as a central regulator of weight, there is considerable interest in its therapeutic potential as a weight loss agent. The small molecule camptothecin exerts anorectic effects in obese mice by inducing the expression of Gdf15 and subsequent activation of GFRAL157. The first phase I clinical trial investigating the therapeutic potential of a long-acting GFRAL receptor agonist has been completed158: in 54 patients with overweight or obesity, weekly subcutaneous injections of LY3463251 induced a 3% decrease in body weight compared with placebo after a 12-week treatment period. This reduction was surprisingly modest compared with the maximal weight reduction of 26% in LY3463251-treated rodents with similar circulating drug concentrations158. LY3463251 also substantially lowered food intake in humans by around 850 kcal per day during ad libitum feeding, indicating that, although GFRAL receptor agonism effectively reduces calorie intake, the weight loss effect is less translatable than expected based on preclinical studies, potentially owing to subtle differences between the synthetic long-acting agonist and endogenous GDF15–GFRAL signalling, a compensatory decrease in energy expenditure, or other experimental design factors such as standardized meals that do not reflect typical eating behaviour151,158.
Future studies are needed to address these issues, to investigate potential similarities and differences in GDF15–GFRAL signalling between humans and animal models, and to elucidate the mechanisms of GDF15 in ORG. In light of evidence of the presence of a bidirectional relationship between the kidney and obesity, treatments that target kidney dysfunction per se might preserve the kidney’s physiological role in regulating body weight. Furthermore, our findings that kidney-derived GDF15 is necessary for metformin-induced weight loss add to the list of kidney-derived messengers, with GDF15 as a secreted factor that enables the kidney to regulate body weight9.
Potential distinct roles for GDF15, SGLT2is and GLP1RAs in mediating weight loss
Combination therapy is often required to attain meaningful weight loss. Greater weight loss is observed in patients treated with metformin and GLP1RA combination therapy than in those treated with metformin monotherapy159, while metformin and dapagliflozin together elicit greater weight loss than either treatment alone in individuals with obesity and metabolic syndrome, suggesting that distinct weight-lowering mechanisms act additively or synergistically160.
GDF15 levels were found to modestly decrease with canagliflozin treatment in individuals in the CANVAS trial161. Canagliflozin conferred a protective effect on cardiorenal outcomes, but this protective effect was not mediated by the decrease in GDF15; the reduction in GDF15 with SGLT2i treatment might be confounded by improvements in chronic disease states, including heart failure.
In rats, the combination of semaglutide and GDF15 treatment also resulted in greater weight loss in comparison with either treatment alone162. However, GDF15 might induce weight loss through mechanisms that are distinct from those induced by GLP1RAs140,162. For example, GLP1 signalling mediates weight loss by increasing the number of IL-6-expressing neurons, whereas GDF15 increases the proportion of calcitonin gene-related peptide neurons163. Semaglutide amplifies satiety induced by cholecystokinin and modulates food motivation, while GDF15 treatment has no effect on feeding motivation or CCK signalling162. In addition, liraglutide maintains its anorectic effect in GDF15-deficient or GFRAL-deficient mice, while GDF15 maintains its anorectic effect in GLP1R-deficient mice140.
A GLP1 and GDF15 dual agonist fusion protein (QL1005) demonstrated superior in vitro pharmacokinetic potency in comparison with semaglutide alone, and induced weight loss, suppressed appetite and improved metabolic parameters in mice. This finding was replicated in monkeys, in which reduction in body weight, suppression of food intake and improvement in glycaemic control and insulin secretion were observed alongside few gastrointestinal adverse effects164. Collectively, future investigations are warranted to explore the potential distinct and/or similar mechanisms of weight loss from SGLT2is, GLP1RAs and GDF15 to evaluate synergistic effects on weight loss.
Conclusions
Obesity alters kidney function through multiple mechanisms, including activation of the RSNS, stimulation of the RAAS, secretion of adipokines, glomerular hyperfiltration, direct renal lipotoxicity, systemic hypertension, and a distinct entity known as ORG. On the other hand, metformin results in the release of GDF15 from the kidney in order to lower food intake and mediate weight loss, thereby establishing a kidney–brain axis in the regulation of energy balance. Our arsenal of therapies for the treatment of obesity, such as bariatric surgery, SGLT2is and GLP1RAs, has expanded rapidly, yet our understanding of the mechanisms of action by which these individual and combined therapies induce weight loss and confer a beneficial impact on kidney function remains incomplete. We propose that kidney function and body weight are related in a bidirectional fashion. Obesity leads to CKD, while the kidney also acts as a key regulator of body weight. Therapies such as SGLT2is, incretin-based therapies and metformin might alter the interrelationship between the kidney and weight to influence human health and disease.
References
NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet 390, 2627–2642 (2017).
Kovesdy, C. P. Epidemiology of chronic kidney disease: an update 2022. Kidney Int. Suppl. 12, 7–11 (2022).
Eckel, R. H. & Cornier, M. A. Update on the NCEP ATP-III emerging cardiometabolic risk factors. BMC Med. 12, 115 (2014).
Ndumele, C. E. et al. Cardiovascular-kidney-metabolic health: a presidential advisory from the American Heart Association. Circulation 148, 1606–1635 (2023).
Tchernof, A. & Després, J. P. Pathophysiology of human visceral obesity: an update. Physiol. Rev. 93, 359–404 (2013).
Ye, C. et al. Causal associations of obesity with chronic kidney disease and arterial stiffness: a mendelian randomization study. J. Clin. Endocrinol. Metab. 107, e825–e835 (2022).
Chang, A. R. et al. Adiposity and risk of decline in glomerular filtration rate: meta-analysis of individual participant data in a global consortium. BMJ 364, k5301 (2019). This meta-analysis demonstrates that increased BMI is an independent risk factor for GFR decline and death in the general population and those with CKD.
Hsu, C. Y., McCulloch, C. E., Iribarren, C., Darbinian, J. & Go, A. S. Body mass index and risk for end-stage renal disease. Ann. Intern. Med. 144, 21–28 (2006).
Zhang, S. Y. et al. Metformin triggers a kidney GDF15-dependent area postrema axis to regulate food intake and body weight. Cell Metab. 35, 875–886.e5 (2023). This study demonstrates that metformin mediates weight loss through a kidney GDF15-dependent area postrema axis.
Weisinger, J. R., Kempson, R. L., Eldridge, F. L. & Swenson, R. S. The nephrotic syndrome: a complication of massive obesity. Ann. Intern. Med. 81, 440–447 (1974).
D’Agati, V. D., Fogo, A. B., Bruijn, J. A. & Jennette, J. C. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am. J. Kidney Dis. 43, 368–382 (2004).
Kambham, N., Markowitz, G. S., Valeri, A. M., Lin, J. & D’Agati, V. D. Obesity-related glomerulopathy: an emerging epidemic. Kidney Int. 59, 1498–1509 (2001).
Chen, H. M. et al. Podocyte lesions in patients with obesity-related glomerulopathy. Am. J. Kidney Dis. 48, 772–779 (2006).
Chen, H. M. et al. Obesity-related glomerulopathy in China: a case series of 90 patients. Am. J. Kidney Dis. 52, 58–65 (2008).
D’Agati, V. D. et al. Obesity-related glomerulopathy: clinical and pathologic characteristics and pathogenesis. Nat. Rev. Nephrol. 12, 453–471 (2016).
Praga, M. et al. Clinical features and long-term outcome of obesity-associated focal segmental glomerulosclerosis. Nephrol. Dial. Transpl. 16, 1790–1798 (2001).
Serra, A. et al. Renal injury in the extremely obese patients with normal renal function. Kidney Int. 73, 947–955 (2008).
Griffin, K. A., Kramer, H. & Bidani, A. K. Adverse renal consequences of obesity. Am. J. Physiol. Ren. Physiol. 294, F685–F696 (2008).
Hall, J. E., do Carmo, J. M., da Silva, A. A., Wang, Z. & Hall, M. E. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ. Res. 116, 991–1006 (2015).
Schorr, U., Blaschke, K., Turan, S., Distler, A. & Sharma, A. M. Relationship between angiotensinogen, leptin and blood pressure levels in young normotensive men. J. Hypertens. 16, 1475–1480 (1998).
Hall, J. E., do Carmo, J. M., da Silva, A. A., Wang, Z. & Hall, M. E. Obesity, kidney dysfunction and hypertension: mechanistic links. Nat. Rev. Nephrol. 15, 367–385 (2019).
Goodfriend, T. L., Ball, D. L., Egan, B. M., Campbell, W. B. & Nithipatikom, K. Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension 43, 358–363 (2004).
Jeon, J. H. et al. A novel adipokine CTRP1 stimulates aldosterone production. FASEB J. 22, 1502–1511 (2008).
Mallamaci, F. et al. ACE inhibition is renoprotective among obese patients with proteinuria. J. Am. Soc. Nephrol. 22, 1122–1128 (2011).
Kim, S. et al. The adipose renin-angiotensin system modulates systemic markers of insulin sensitivity and activates the intrarenal renin-angiotensin system. J. Biomed. Biotechnol. 2006, 27012 (2006).
Henegar, J. R. et al. Catheter-based radiorefrequency renal denervation lowers blood pressure in obese hypertensive dogs. Am. J. Hypertens. 27, 1285–1292 (2014).
Xu, X., Huang, X., Zhang, L., Qin, Z. & Hua, F. Adiponectin protects obesity-related glomerulopathy by inhibiting ROS/NF-κB/NLRP3 inflammation pathway. BMC Nephrol. 22, 218 (2021).
Hall, J. E. et al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J. Biol. Chem. 285, 17271–17276 (2010).
Mansukhani, M. P., Wang, S. & Somers, V. K. Chemoreflex physiology and implications for sleep apnoea: insights from studies in humans. Exp. Physiol. 100, 130–135 (2015).
Hall, M. E. et al. Obesity, hypertension, and chronic kidney disease. Int. J. Nephrol. Renovasc Dis. 7, 75–88 (2014).
Eddy, A. A. & Fogo, A. B. Plasminogen activator inhibitor-1 in chronic kidney disease: evidence and mechanisms of action. J. Am. Soc. Nephrol. 17, 2999–3012 (2006).
Benomar, Y. et al. Central resistin overexposure induces insulin resistance through Toll-like receptor 4. Diabetes 62, 102–114 (2013).
Yano, Y. et al. Differential impacts of adiponectin on low-grade albuminuria between obese and nonobese persons without diabetes. J. Clin. Hypertens. 9, 775–782 (2007).
Sharma, K. et al. Adiponectin regulates albuminuria and podocyte function in mice. J. Clin. Invest. 118, 1645–1656 (2008).
Fang, F. et al. Deletion of the gene for adiponectin accelerates diabetic nephropathy in the Ins2+/C96Y mouse. Diabetologia 58, 1668–1678 (2015).
Ohashi, K. et al. Exacerbation of albuminuria and renal fibrosis in subtotal renal ablation model of adiponectin-knockout mice. Arterioscler. Thromb. Vasc. Biol. 27, 1910–1917 (2007).
Briffa, J. F., McAinch, A. J., Poronnik, P. & Hryciw, D. H. Adipokines as a link between obesity and chronic kidney disease. Am. J. Physiol. Ren. Physiol. 305, F1629–F1636 (2013).
Aizawa-Abe, M. et al. Pathophysiological role of leptin in obesity-related hypertension. J. Clin. Invest. 105, 1243–1252 (2000).
Huby, A. C. et al. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation 132, 2134–2145 (2015).
Belin de Chantemèle, E. J., Mintz, J. D., Rainey, W. E. & Stepp, D. W. Impact of leptin-mediated sympatho-activation on cardiovascular function in obese mice. Hypertension 58, 271–279 (2011).
de Vries, A. P. et al. Fatty kidney: emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2, 417–426 (2014).
Zhao, J. et al. CD36-mediated lipid accumulation and activation of NLRP3 inflammasome lead to podocyte injury in obesity-related glomerulopathy. Mediators Inflamm. 2019, 3172647 (2019).
Shahzad, K. et al. Podocyte-specific Nlrp3 inflammasome activation promotes diabetic kidney disease. Kidney Int. 102, 766–779 (2022).
Hou, Y. et al. NLRP3 inflammasome negatively regulates podocyte autophagy in diabetic nephropathy. Biochem. Biophys. Res. Commun. 521, 791–798 (2020).
Qiu, Y. Y. & Tang, L. Q. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol. Res. 114, 251–264 (2016).
Ke, B., Shen, W., Fang, X. & Wu, Q. The NLPR3 inflammasome and obesity-related kidney disease. J. Cell Mol. Med. 22, 16–24 (2018).
Rampanelli, E. et al. Metabolic injury-induced NLRP3 inflammasome activation dampens phospholipid degradation. Sci. Rep. 7, 2861 (2017).
Yamamoto, T. et al. High-fat diet-induced lysosomal dysfunction and impaired autophagic flux contribute to lipotoxicity in the kidney. J. Am. Soc. Nephrol. 28, 1534–1551 (2017).
Bakker, P. J. et al. Nlrp3 is a key modulator of diet-induced nephropathy and renal cholesterol accumulation. Kidney Int. 85, 1112–1122 (2014).
Chiazza, F. et al. Targeting the NLRP3 inflammasome to reduce diet-induced metabolic abnormalities in mice. Mol. Med. 21, 1025–1037 (2016).
The Diabetes Prevention Program Research Group Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care 35, 731–737 (2012).
Seifarth, C., Schehler, B. & Schneider, H. J. Effectiveness of metformin on weight loss in non-diabetic individuals with obesity. Exp. Clin. Endocrinol. Diabetes 121, 27–31 (2013).
Sjöström, L. et al. Effects of bariatric surgery on mortality in Swedish obese subjects. N. Engl. J. Med. 357, 741–752 (2007).
Alexander, J. W., Goodman, H. R., Hawver, L. R. & Cardi, M. A. Improvement and stabilization of chronic kidney disease after gastric bypass. Surg. Obes. Relat. Dis. 5, 237–241 (2009).
Bilha, S. C. et al. The effects of bariatric surgery on renal outcomes: a systematic review and meta-analysis. Obes. Surg. 28, 3815–3833 (2018).
Inge, T. H. et al. Weight loss and health status 3 years after bariatric surgery in adolescents. N. Engl. J. Med. 374, 113–123 (2016).
Schiavon, C. A. et al. Effects of bariatric surgery in obese patients with hypertension: the GATEWAY randomized trial (Gastric Bypass to Treat Obese Patients With Steady Hypertension). Circulation 137, 1132–1142 (2018).
Scheurlen, K. M. et al. Metabolic surgery improves renal injury independent of weight loss: a meta-analysis. Surg. Obes. Relat. Dis. 15, 1006–1020 (2019).
MacLaughlin, H. L. et al. Weight loss, adipokines, and quality of life after sleeve gastrectomy in obese patients with stages 3-4 CKD: a randomized controlled pilot study. Am. J. Kidney Dis. 64, 660–663 (2014).
Haynes, W. G., Morgan, D. A., Walsh, S. A., Mark, A. L. & Sivitz, W. I. Receptor-mediated regional sympathetic nerve activation by leptin. J. Clin. Invest. 100, 270–278 (1997).
Moritz, E. et al. Renal sinus fat is expanded in patients with obesity and/or hypertension and reduced by bariatric surgery associated with hypertension remission. Metabolites 12, 617 (2022).
Ricci, M. A. et al. Morbid obesity and hypertension: the role of perirenal fat. J. Clin. Hypertens. 20, 1430–1437 (2018).
Docherty, N. G. & le Roux, C. W. Bariatric surgery for the treatment of chronic kidney disease in obesity and type 2 diabetes mellitus. Nat. Rev. Nephrol. 16, 709–720 (2020).
Ghezzi, C., Loo, D. D. F. & Wright, E. M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 61, 2087–2097 (2018).
Shyangdan, D. S., Uthman, O. A. & Waugh, N. SGLT-2 receptor inhibitors for treating patients with type 2 diabetes mellitus: a systematic review and network meta-analysis. BMJ Open. 6, e009417 (2016).
Herrington, W. G. et al. Empagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 388, 117–127 (2023).
Perkovic, V. et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N. Engl. J. Med. 380, 2295–2306 (2019).
Cherney, D. Z. et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 129, 587–597 (2014).
van Bommel, E. J. M. et al. The renal hemodynamic effects of the SGLT2 inhibitor dapagliflozin are caused by post-glomerular vasodilatation rather than pre-glomerular vasoconstriction in metformin-treated patients with type 2 diabetes in the randomized, double-blind RED trial. Kidney Int. 97, 202–212 (2020).
Körner, A., Eklöf, A. C., Celsi, G. & Aperia, A. Increased renal metabolism in diabetes. Mechanism and functional implications. Diabetes 43, 629–633 (1994).
Vallon, V. & Thomson, S. C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 16, 317–336 (2020).
Ferrannini, E., Mark, M. & Mayoux, E. CV protection in the EMPA-REG OUTCOME trial: a “thrifty substrate” hypothesis. Diabetes Care 39, 1108–1114 (2016).
Heerspink, H. J. L. et al. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 383, 1436–1446 (2020).
Mayne, K. J. et al. Effects of empagliflozin on fluid overload, weight and blood pressure in chronic kidney disease. J. Am. Soc. Nephrol. https://doi.org/10.1681/ASN.0000000000000271 (2023).
Yau, K., Dharia, A., Alrowiyti, I. & Cherney, D. Z. I. Prescribing SGLT2 inhibitors in patients with CKD: expanding indications and practical considerations. Kidney Int. Rep. 7, 1463–1476 (2022).
Thomas, M. C. & Cherney, D. Z. I. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 61, 2098–2107 (2018).
Horie, I. et al. Increased sugar intake as a form of compensatory hyperphagia in patients with type 2 diabetes under dapagliflozin treatment. Diabetes Res. Clin. Pract. 135, 178–184 (2018).
Ferrannini, G. et al. Energy balance after sodium-glucose cotransporter 2 inhibition. Diabetes Care 38, 1730–1735 (2015).
Rajeev, S. P. et al. No evidence of compensatory changes in energy balance, despite reductions in body weight and liver fat, during dapagliflozin treatment in type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled, cross-over trial (ENERGIZE). Diabetes Obes. Metab. 25, 3621–3631 (2023).
Sargeant, J. A. et al. The effects of empagliflozin, dietary energy restriction, or both on appetite-regulatory gut peptides in individuals with type 2 diabetes and overweight or obesity: the SEESAW randomized, double-blind, placebo-controlled trial. Diabetes Obes. Metab. 24, 1509–1521 (2022).
Cefalu, W. T. Paradoxical insights into whole body metabolic adaptations following SGLT2 inhibition. J. Clin. Invest. 124, 485–487 (2014).
Ansary, T. M., Nakano, D. & Nishiyama, A. Diuretic effects of sodium glucose cotransporter 2 inhibitors and their influence on the renin-angiotensin system. Int. J. Mol. Sci. 20, 629 (2019).
Marton, A. et al. Organ protection by SGLT2 inhibitors: role of metabolic energy and water conservation. Nat. Rev. Nephrol. 17, 65–77 (2021).
Yasui, A. et al. Empagliflozin induces transient diuresis without changing long-term overall fluid balance in Japanese patients with type 2 diabetes. Diabetes Ther. 9, 863–871 (2018).
Wild, J. et al. Aestivation motifs explain hypertension and muscle mass loss in mice with psoriatic skin barrier defect. Acta Physiol. 232, e13628 (2021).
Kitada, K. et al. High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J. Clin. Invest. 127, 1944–1959 (2017).
Cherney, D. Z. I. et al. Pooled analysis of phase III trials indicate contrasting influences of renal function on blood pressure, body weight, and HbA1c reductions with empagliflozin. Kidney Int. 93, 231–244 (2018).
DeFronzo, R. A., Reeves, W. B. & Awad, A. S. Pathophysiology of diabetic kidney disease: impact of SGLT2 inhibitors. Nat. Rev. Nephrol. 17, 319–334 (2021).
Xu, L. et al. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine 20, 137–149 (2017).
Tomita, I. et al. SGLT2 inhibition mediates protection from diabetic kidney disease by promoting ketone body-induced mTORC1 inhibition. Cell Metab. 32, 404–419.e6 (2020). This study demonstrates that SGLT2 inhibitors increase ketone bodies to correct mTORC1 overactivation to mediate kidney protection in DKD.
Holst, J. J., Orskov, C., Nielsen, O. V. & Schwartz, T. W. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett. 211, 169–174 (1987).
Cherney, D. Z. I., Udell, J. A. & Drucker, D. J. Cardiorenal mechanisms of action of glucagon-like-peptide-1 receptor agonists and sodium-glucose cotransporter 2 inhibitors. Med 2, 1203–1230 (2021).
Kristensen, S. L. et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 7, 776–785 (2019).
Shi, Q. et al. Pharmacotherapy for adults with overweight and obesity: a systematic review and network meta-analysis of randomised controlled trials. Lancet 399, 259–269 (2022).
Sisley, S. et al. Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effect. J. Clin. Invest. 124, 2456–2463 (2014).
Heiss, C. N. et al. The gut microbiota regulates hypothalamic inflammation and leptin sensitivity in Western diet-fed mice via a GLP-1R-dependent mechanism. Cell Rep. 35, 109163 (2021).
Drucker, D. J. GLP-1 physiology informs the pharmacotherapy of obesity. Mol. Metab. 57, 101351 (2022).
Turton, M. D. et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 379, 69–72 (1996).
Ludwig, M. Q. et al. A genetic map of the mouse dorsal vagal complex and its role in obesity. Nat. Metab. 3, 530–545 (2021).
Secher, A. et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J. Clin. Invest. 124, 4473–4488 (2014).
Fortin, S. M. et al. GABA neurons in the nucleus tractus solitarius express GLP-1 receptors and mediate anorectic effects of liraglutide in rats. Sci. Transl. Med. 12, eaay8071 (2020).
Nauck, M. A., Quast, D. R., Wefers, J. & Meier, J. J. GLP-1 receptor agonists in the treatment of type 2 diabetes – state-of-the-art. Mol. Metab. 46, 101102 (2021).
Sattar, N. et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 9, 653–662 (2021).
Yau, K., Odutayo, A., Dash, S. & Cherney, D. Z. I. Biology and clinical use of glucagon-like-peptide-1 receptor agonists in vascular protection. Can. J. Cardiol. 39, 1816–1838 (2023).
Kawanami, D. & Takashi, Y. GLP-1 receptor agonists in diabetic kidney disease: from clinical outcomes to mechanisms. Front. Pharmacol. 11, 967 (2020).
Górriz, J. L. et al. GLP-1 receptor agonists and diabetic kidney disease: a call of attention to nephrologists. J. Clin. Med. 9, 947 (2020).
Gutzwiller, J. P. et al. Glucagon-like peptide 1 induces natriuresis in healthy subjects and in insulin-resistant obese men. J. Clin. Endocrinol. Metab. 89, 3055–3061 (2004).
Tonneijck, L. et al. Acute renal effects of the GLP-1 receptor agonist exenatide in overweight type 2 diabetes patients: a randomised, double-blind, placebo-controlled trial. Diabetologia 59, 1412–1421 (2016).
Rossing, P. et al. The rationale, design and baseline data of FLOW, a kidney outcomes trial with once-weekly semaglutide in people with type 2 diabetes and chronic kidney disease. Nephrol. Dial. Transpl. 38, 2041–2051 (2023).
Willard, F. S. et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight 5, e140532 (2020).
Heerspink, H. J. L. et al. Effects of tirzepatide versus insulin glargine on kidney outcomes in type 2 diabetes in the SURPASS-4 trial: post-hoc analysis of an open-label, randomised, phase 3 trial. Lancet Diabetes Endocrinol. 10, 774–785 (2022).
Sánchez-Garrido, M. A. et al. GLP-1/glucagon receptor co-agonism for treatment of obesity. Diabetologia 60, 1851–1861 (2017).
Jastreboff, A. M. et al. Triple-hormone-receptor agonist retatrutide for obesity – a phase 2 trial. N. Engl. J. Med. 389, 514–526 (2023).
Kaneko, K. et al. Gut-derived GIP activates central Rap1 to impair neural leptin sensitivity during overnutrition. J. Clin. Invest. 129, 3786–3791 (2019).
Killion, E. A. et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci. Transl. Med. 10, eaat3392 (2018).
Zhang, Q. et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. 33, 833–844.e5 (2021).
Adriaenssens, A. E. et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab. 30, 987–996.e6 (2019).
Quiñones, M. et al. Hypothalamic CaMKKβ mediates glucagon anorectic effect and its diet-induced resistance. Mol. Metab. 4, 961–970 (2015).
Lu, S. C. et al. GIPR antagonist antibodies conjugated to GLP-1 peptide are bispecific molecules that decrease weight in obese mice and monkeys. Cell Rep. Med. 2, 100263 (2021).
Boyle, C. N., Lutz, T. A. & Le Foll, C. Amylin – its role in the homeostatic and hedonic control of eating and recent developments of amylin analogs to treat obesity. Mol. Metab. 8, 203–210 (2018).
Enebo, L. B. et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2·4 mg for weight management: a randomised, controlled, phase 1b trial. Lancet 397, 1736–1748 (2021).
Chen, W., Binbin, G., Lidan, S., Qiang, Z. & Jing, H. Evolution of peptide YY analogs for the management of type 2 diabetes and obesity. Bioorg. Chem. 140, 106808 (2023).
Hall, M. E., Jordan, J. H., Juncos, L. A., Hundley, W. G. & Hall, J. E. BOLD magnetic resonance imaging in nephrology. Int. J. Nephrol. Renovasc Dis. 11, 103–112 (2018).
Miller, R. A. et al. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 494, 256–260 (2013).
Day, E. A. et al. Metformin-induced increases in GDF15 are important for suppressing appetite and promoting weight loss. Nat. Metab. 1, 1202–1208 (2019).
Coll, A. P. et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature 578, 444–448 (2020). This study demonstrated that metformin increases circulating GDF15, which is necessary for its effects on energy balance and body weight.
Bootcov, M. R. et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-β superfamily. Proc. Natl Acad. Sci. USA 94, 11514–11519 (1997).
Lawton, L. N. et al. Identification of a novel member of the TGF-beta superfamily highly expressed in human placenta. Gene 203, 17–26 (1997).
Böttner, M., Suter-Crazzolara, C., Schober, A. & Unsicker, K. Expression of a novel member of the TGF-β superfamily, growth/differentiation factor-15/macrophage-inhibiting cytokine-1 (GDF-15/MIC-1) in adult rat tissues. Cell Tissue Res. 297, 103–110 (1999).
Patel, S. et al. GDF15 provides an endocrine signal of nutritional stress in mice and humans. Cell Metab. 29, 707–718.e8 (2019).
Wang, D. et al. GDF15: emerging biology and therapeutic applications for obesity and cardiometabolic disease. Nat. Rev. Endocrinol. 17, 592–607 (2021).
Hsu, J. Y. et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 550, 255–259 (2017).
Yang, L. et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 23, 1158–1166 (2017).
Mullican, S. E. et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 23, 1150–1157 (2017).
Emmerson, P. J. et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 23, 1215–1219 (2017).
Wang, D. et al. GDF15 promotes weight loss by enhancing energy expenditure in muscle. Nature 619, 143–150 (2023).
Xiong, Y. et al. Long-acting MIC-1/GDF15 molecules to treat obesity: evidence from mice to monkeys. Sci. Transl. Med. 9, eaan8732 (2017).
Sabatini, P. V. et al. GFRAL-expressing neurons suppress food intake via aversive pathways. Proc. Natl Acad. Sci. USA 118, e2021357118 (2021).
Blundell, J. et al. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes. Metab. 19, 1242–1251 (2017).
Frikke-Schmidt, H. et al. GDF15 acts synergistically with liraglutide but is not necessary for the weight loss induced by bariatric surgery in mice. Mol. Metab. 21, 13–21 (2019).
Chrysovergis, K. et al. NAG-1/GDF-15 prevents obesity by increasing thermogenesis, lipolysis and oxidative metabolism. Int. J. Obes. 38, 1555–1564 (2014).
Gerstein, H. C. et al. Growth differentiation factor 15 as a novel biomarker for metformin. Diabetes Care 40, 280–283 (2017).
Kwon, S. et al. The long-term effects of metformin on patients with type 2 diabetic kidney disease. Diabetes Care 43, 948–955 (2020).
Lee, M. et al. Phosphorylation of acetyl-CoA carboxylase by AMPK reduces renal fibrosis and is essential for the anti-fibrotic effect of metformin. J. Am. Soc. Nephrol. 29, 2326–2336 (2018).
Han, Y. C. et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis. 12, 925 (2021).
Christensen, M. et al. Renoprotective effects of metformin are independent of organic cation transporters 1 & 2 and AMP-activated protein kinase in the kidney. Sci. Rep. 6, 35952 (2016).
Mazagova, M. et al. Genetic deletion of growth differentiation factor 15 augments renal damage in both type 1 and type 2 models of diabetes. Am. J. Physiol. Ren. Physiol. 305, F1249–F1264 (2013).
Valiño-Rivas, L. et al. Growth differentiation factor-15 preserves Klotho expression in acute kidney injury and kidney fibrosis. Kidney Int. 101, 1200–1215 (2022).
Zhou, Z. et al. Circulating GDF-15 in relation to the progression and prognosis of chronic kidney disease: a systematic review and dose-response meta-analysis. Eur. J. Intern. Med. 110, 77–85 (2023).
Nair, V. et al. Growth differentiation factor-15 and risk of CKD progression. J. Am. Soc. Nephrol. 28, 2233–2240 (2017).
Breit, S. N., Brown, D. A. & Tsai, V. W. W. GDF15 analogs as obesity therapeutics. Cell Metab. 35, 227–228 (2023).
Connelly, P. W. et al. Growth differentiation factor 15 is decreased by kidney transplantation. Clin. Biochem. 73, 57–61 (2019).
Seyfried, F. et al. Weight loss from caloric restriction vs Roux-en-Y gastric bypass surgery differentially regulates systemic and portal vein GDF15 levels in obese Zucker fatty rats. Physiol. Behav. 240, 113534 (2021).
Dolo, P. R. et al. Effect of sleeve gastrectomy on plasma growth differentiation factor-15 (GDF15) in human. Am. J. Surg. 220, 725–730 (2020).
Salman, A. et al. Changes in plasma growth differentiation factor-15 after laparoscopic sleeve gastrectomy in morbidly obese patients: a prospective study. J. Inflamm. Res. 14, 1365–1373 (2021).
Kleinert, M. et al. Effect of bariatric surgery on plasma GDF15 in humans. Am. J. Physiol. Endocrinol. Metab. 316, E615–E621 (2019).
Lu, J. F. et al. Camptothecin effectively treats obesity in mice through GDF15 induction. PLoS Biol. 20, e3001517 (2022).
Benichou, O. et al. Discovery, development, and clinical proof of mechanism of LY3463251, a long-acting GDF15 receptor agonist. Cell Metab. 35, 274–286.e10 (2023).
Lyu, X. et al. The antiobesity effect of GLP-1 receptor agonists alone or in combination with metformin in overweight/obese women with polycystic ovary syndrome: a systematic review and meta-analysis. Int. J. Endocrinol. 2021, 6616693 (2021).
Cheng, L. et al. Dapagliflozin, metformin, monotherapy or both in patients with metabolic syndrome. Sci. Rep. 11, 24263 (2021).
Sen, T. et al. Association between circulating GDF-15 and cardio-renal outcomes and effect of canagliflozin: results from the CANVAS trial. J. Am. Heart Assoc. 10, e021661 (2021).
Ghidewon, M. et al. Growth differentiation factor 15 (GDF15) and semaglutide inhibit food intake and body weight through largely distinct, additive mechanisms. Diabetes Obes. Metab. 24, 1010–1020 (2022).
Anesten, F. et al. Glucagon-like peptide-1-, but not growth and differentiation factor 15-, receptor activation increases the number of interleukin-6-expressing cells in the external lateral parabrachial nucleus. Neuroendocrinology 109, 310–321 (2019).
Zhang, Y. et al. Activity-balanced GLP-1/GDF15 dual agonist reduces body weight and metabolic disorder in mice and non-human primates. Cell Metab. 35, 287–298.e4 (2023).
Jastreboff, A. M. et al. Tirzepatide once weekly for the treatment of obesity. N. Engl. J. Med. 387, 205–216 (2022).
Garvey, W. T. et al. Tirzepatide once weekly for the treatment of obesity in people with type 2 diabetes (SURMOUNT-2): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 402, 613–626 (2023).
Parker, V. E. R. et al. Efficacy and safety of cotadutide, a dual glucagon-like peptide-1 and glucagon receptor agonist, in a randomized phase 2a study of patients with type 2 diabetes and chronic kidney disease. Diabetes Obes. Metab. 24, 1360–1369 (2022).
Jungnik, A. et al. Phase I studies of the safety, tolerability, pharmacokinetics and pharmacodynamics of the dual glucagon receptor/glucagon-like peptide-1 receptor agonist BI 456906. Diabetes Obes. Metab. 25, 1011–1023 (2023).
Romero-Gómez, M. et al. A phase 2a active-comparator-controlled study to evaluate the efficacy and safety of efinopegdutide in patients with nonalcoholic fatty liver disease. J. Hepatol. 79, 888–897 (2023).
Rosenstock, J. et al. Retatrutide, a GIP, GLP-1 and glucagon receptor agonist, for people with type 2 diabetes: a randomised, double-blind, placebo and active-controlled, parallel-group, phase 2 trial conducted in the USA. Lancet 402, 529–544 (2023).
Urva, S. et al. LY3437943, a novel triple GIP, GLP-1, and glucagon receptor agonist in people with type 2 diabetes: a phase 1b, multicentre, double-blind, placebo-controlled, randomised, multiple-ascending dose trial. Lancet 400, 1869–1881 (2022).
Bossart, M. et al. Effects on weight loss and glycemic control with SAR441255, a potent unimolecular peptide GLP-1/GIP/GCG receptor triagonist. Cell Metab. 34, 59–74.e10 (2022).
Acknowledgements
K.Y. is supported by the University of Toronto Department of Medicine Eliot Phillipson Clinician Scientist Training Program, Clarence Henry Trelford Clinician Scientist Award in Diabetes, and a KRESCENT Postdoctoral Fellowship. The KRESCENT program is co-sponsored by the Kidney Foundation of Canada, the Canadian Society of Nephrology, and the Canadian Institute of Health Research (CIHR). R.K. is supported by a CIHR graduate award and a Banting and Best Diabetes Centre graduate scholarship. D.Z.I.C. is supported by a Department of Medicine, University of Toronto Merit Award, and receives support from the CIHR, Diabetes Canada and the Heart and Stroke Richard Lewar Centre of Excellence. D.Z.I.C. is also the recipient of a CIHR-KFOC Teams Grant Award. T.K.T.L. is supported by CIHR grants (PJT 189957, PJT 183901, MRT-168045) and holds the Canada Research Chair (Tier 1) in Diabetes and Obesity at the Toronto General Hospital Research Institute & University of Toronto.
Author information
Authors and Affiliations
Contributions
All authors researched data for the article, contributed substantially to discussion of content, wrote the manuscript and reviewed and edited the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
D.Z.I.C. has received consulting fees or speaking honoraria or both from Janssen, Bayer, Boehringer Ingelheim-Eli, Lilly, AstraZeneca, Merck & Co Inc, Prometic and Sanofi, and has received operating funds from Janssen, Boehringer Ingelheim-Eli, Lilly, Sanofi, AstraZeneca and Merck & Co Inc. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Endocrinology thanks Alex R. Chang, Neil Docherty and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yau, K., Kuah, R., Cherney, D.Z.I. et al. Obesity and the kidney: mechanistic links and therapeutic advances. Nat Rev Endocrinol (2024). https://doi.org/10.1038/s41574-024-00951-7
Accepted:
Published:
DOI: https://doi.org/10.1038/s41574-024-00951-7
