
Abstract
The complement system is a recognized pillar of host defence against infection and noxious self-derived antigens. Complement is traditionally known as a serum-effective system, whereby the liver expresses and secretes most complement components, which participate in the detection of bloodborne pathogens and drive an inflammatory reaction to safely remove the microbial or antigenic threat. However, perturbations in normal complement function can cause severe disease and, for reasons that are currently not fully understood, the kidney is particularly vulnerable to dysregulated complement activity. Novel insights into complement biology have identified cell-autonomous and intracellularly active complement — the complosome — as an unexpected central orchestrator of normal cell physiology. For example, the complosome controls mitochondrial activity, glycolysis, oxidative phosphorylation, cell survival and gene regulation in innate and adaptive immune cells, and in non-immune cells, such as fibroblasts and endothelial and epithelial cells. These unanticipated complosome contributions to basic cell physiological pathways make it a novel and central player in the control of cell homeostasis and effector responses. This discovery, together with the realization that an increasing number of human diseases involve complement perturbations, has renewed interest in the complement system and its therapeutic targeting. Here, we summarize the current knowledge about the complosome across healthy cells and tissues, highlight contributions from dysregulated complosome activities to human disease and discuss potential therapeutic implications.
Key points
-
Complement function is compartmentalized and operates systemically, locally in the extracellular space, and intracellularly within sub-cellular compartments and organelles.
-
Intracellular complement — the complosome — serves non-classical roles as a novel central regulator of basic cell physiological processes including mitochondrial respiration, glycolysis, autophagy and gene transcription.
-
The complosome functions across immune and non-immune cells and tissues where it controls normal cell turnover, the responses to infectious and non-infectious stimuli and the return to homeostasis.
-
Perturbations in complosome activities contribute to human disease, including infections and infection-related pathological conditions, arthritic disease, atherosclerosis, cancer and kidney disease.
-
Targeting the complosome, possibly in combination with the extracellularly active complement, might be therapeutically beneficial in complement-mediated pathological conditions.
Similar content being viewed by others
Introduction
The complement system was discovered over a century ago by Jules Bordet as a serum-operative key arm of innate immunity that ‘complemented’ the activity of antibodies during the detection and removal of bloodborne pathogens1. Complement soon became known as a formidable killer, as it was the force behind the direct lytic destruction of dangerous microbes2. As the structures and functions of single complement components were elucidated, the system became recognized as a mediator of the acute inflammatory reaction with important roles in directing cell migration and activation of innate immune cells2,3. However, work that begun in the 1980s and is still ongoing demonstrated that complement receptor signalling on B and T lymphocytes also constitutes an unexpected integral part of humoral and adaptive cellular immune responses4,5,6. We are now on the verge of a similar fundamental change in our understanding of complement. Complement activation and effector functions are more compartmentalized than previously thought and occur not only extracellularly, but also intracellularly across a broad range of cell populations and tissues. This intracellularly active complement system was coined ‘the complosome’7,8. Ongoing work on the complosome continues to unveil strong and exciting evidence that complement activity in this new location might have novel and non-canonical functions, as the complosome actively participates in basic cellular processes, such as metabolism, autophagy and gene expression9,10,11. In line with its substantial role in the regulation of normal cell biology, perturbations in complosome activities are associated with several prevalent human diseases, including recurrent infections, arthritic disease, atherosclerosis and cancer12,13. Interestingly, emerging data suggest that dysregulation of intracellularly active complement might have a prominent role in the kidney, which is frequently affected by complement deposition14, often for reasons that are not fully understood.
In this Review, we discuss the relatively new concept of the complosome7,8 and examine current evidence about the emerging roles of intracellular complement activity across healthy immune and non-immune cells in physiological cellular processes. Further, we focus on the known contributions of the complosome to human disease, including kidney disease. Finally, we consider the therapeutic potential of targeting the complosome in complement-mediated pathologies and highlight key limitations and caveats of complosome-focused clinical approaches.
Classical functions of systemic and local extracellular complement
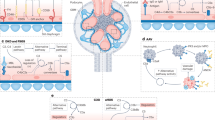
The human complement system comprises >50 proteins that either circulate in blood (specifically, core components and some complement regulators) or lymph, or exist as cell membrane-bound proteins (regulators and receptors)1,2. C3 and C5 are major effector molecules that are mostly secreted by the liver in a pro-enzymatic form. Complement C3 and C5 activation is initiated when one or several activation pathways is triggered by pathogen- or damage-associated molecular patterns (Fig. 1a). This recognition leads to the formation of C3 and C5 convertases, which then cleavage-activate C3 into C3a and C3b, and C5 into C5a and C5b, respectively. C5b combines with serum C6–C9 to form the membrane attack complex (MAC), which induces direct lytic killing of pathogens or noxious target cells. C3b opsonizes microbes and noxious host cells, which induces scavenger cells to phagocytose and destroy C3b-tagged targets1,2,15,16. Receptors for the anaphylatoxins C3a and C5a — C3a receptor (C3aR) and C5aR1 and 2, respectively — are expressed by most host immune and non-immune cells17,18,19. Stimulation of these receptors induces a range of responses, including activation of the endothelium to support adherence and tissue influx of immune cells, smooth muscle cell contraction, and migration and activation of innate immune cells. All of these events underlie the classic inflammatory reaction2,3,20 and are widely considered to be canonical complement functions. The importance of liver-derived circulating complement to the detection and containment of bloodborne pathogens is underpinned by the recurrent bacterial infections that affect individuals with deficiencies in either C3 or C5 (refs. 21,22). Of note, unwanted or chronic complement activation causes detrimental pathological tissue conditions23,24 and the system is therefore tightly controlled by a range of fluid-phase and cell-expressed regulators25,26 (Fig. 1a).

a, Circulating liver-produced complement can be activated through three pathways that result in the formation of C3 and C5 convertases, which cleavage-activate C3 into C3a and C3b, and C5 into C5a and C5b, respectively. This process leads to the formation of the membrane attack complex (MAC) and the induction of classical complement functions. Several regulators (in red) control complement activation. b, Systemic complement protects against bloodborne threats, whereas extra-hepatic cell-derived local complement activation, driven by C3 and C5 secreted by immune and non-immune cells (and subsequent extracellular formation of C3 and/or C5 convertase formation) supports cell survival and cell-specific effector responses in an autocrine and/or paracrine (not shown) manner. c, Activation of cell-autonomous, intracellular complement in immune and non-immune cells can occur at different subcellular locations. The generated activation fragments perform their activities across distinct cell sub-compartments and support normal physiological processes. CD46 is included because its cleaved intracellular domain is considered to be a member of the complosome. ATP prod., adenosine triphosphate production; C1-INH, C1 and MASP1/2 inhibitor; C3aR, C3a receptor; C4BP, C4b binding protein; C5aR, C5a receptor; CR1, complement receptor 1; ER, endoplasmic reticulum; ETC, electron transport chain; F, factor; MAC, membrane attack complex; MASP1/2, MBL serine proteases 1 and 2; MAVS, mitochondrial anti-viral signalling protein; MBL, mannose-binding lectin; mTOR, mammalian target of rapamycin; mTORC1, mammalian target of rapamycin complex 1; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3 inflammasome; OXPHOS, oxidative phosphorylation; PW, pathways; ROS, reactive oxygen species. *Although hepatocytes are the principal source of secreted and circulating C3 and C5, they also engage cell-intrinsic intracellular C3 for homeostatic control of their lipid metabolism.
Although complement is traditionally known as a central arm of innate immunity, it has equally important roles in the regulation of adaptive immunity. For example, the iC3b/C3dg/C3d-binding complement receptor 2 (CR2; also known as CD21) is an important co-stimulatory molecule for B cells and lowers the threshold of B cell receptor (BCR) signalling by up to 10,000-fold4,6,27,28. Similarly, the complement regulator and receptor CD46 (which binds and inactivates C3b and C4b) provides co-stimulatory signals to human CD4+ T cells that are required for normal induction of T helper 1 (TH1) cell responses29,30,31. The discovery that, similar to Toll-like receptors (TLRs), complement is an integral part of adaptive immunity explains the broad impact of complement dysregulation across innate and adaptive immunopathology-mediated human disease states32,33.
Interestingly, studies on the role of complement in adaptive immunity revealed that engagement of complement receptors on immune cells is largely independent of circulating complement and, instead, is mostly driven by immune cell-derived local complement production and its extracellular activation (Fig. 1b). For example, C3 and C5 secreted by activated antigen-presenting cells and/or T cells during cognate antigen-presenting cell–T cell interactions are activated in the extracellular space by proteases or C3/C5 convertases (with antigen-presenting cells also being a source of required local factor D (FD) and FB generation)34,35,36,37. The anaphylatoxins C3a and C5a then engage their receptors expressed on neighbouring cells in an autocrine and/or paracrine manner, and induce specific cellular responses (for example, expression of co-stimulatory molecules, cell proliferation and cytokine production). Pathological changes in local cell-intrinsic complement activities are associated with infections, autoimmunity, transplant rejection and kidney injury35,36,37,38,39,40,41. Current evidence therefore suggests a division of labour in the complement system, whereby circulating complement guards the vascular space through its canonical antimicrobial functions, whereas local cell-derived canonical complement regulates the behaviour of cells (including immune cells) (Fig. 1b).
Intracellular complement and its role in cell physiology
Over the past decade, several studies have shown that the compartmentalization of complement function extends to a third location — the interior of cells (Fig. 1). We have used the term ‘complosome’ to refer to this high-order protein complex (‘-some’), that is, active intracellularly, in contrast to extracellular complement, and whose components not only interact with each other but also with other intracellular danger sensor and effector systems (for example, inflammasomes42,43, autophagosomes44 and the ribosomal machinery45). Complosome proteins are encoded by the same genes that give rise to the liver-derived circulating complement system. Furthermore, intracellular C3 and C5 can also be cleavage-activated by specific proteases8,46 or by intracellular C3 and C5 convertases that form beneath the plasma membrane and on the surface of subcellular compartments43. Of note, although complosome components derive from cell-intrinsic expression in most cells, intracellular C3 can also be sourced from the extracellular space or the cell surface to become part of the complosome that is functionally active intracellularly (Box 1). Complement components produced by immune cells but then secreted to function in an autocrine or paracrine fashion on the cell surface (see above) fall somewhat into a grey zone with regard to complosome definition. We suggest describing such complement activities as ‘cell-autonomous and extracellular’.
The core complosome components C3 and C5, and/or their activation fragments and receptors, as well as some complement regulators have been detected in the cytoplasm, lysosome, endoplasmic reticulum, outer membrane of mitochondria and the nucleus. Importantly, these distinct intracellular locations enable functional roles that differ from those associated with classical complement. Specifically, the complosome participates in basic cell physiological processes including cell metabolism9, autophagy11 and regulation of gene expression10 (Fig. 1c). Intracellular mitochondrial, lysosomal, and/or endosomal C3aR and C5aR1 exert their function via the induction of signalling events43,46,47,48 akin to those triggered by engagement of their ‘classical complement’ counterparts that are expressed on the cell surface. However, we largely consider the functions of the complosome to be non-canonical owing to its unique ability to engage directly with the basic physiology-sustaining cell machinery, which is spatially separated from extracellular complement components (Fig. 1c).
Cell metabolism
Metabolism control seems to represent one of the major functions of the complosome (as has previously been reviewed7,49,50,51,52; here, we only provide a succinct overview). Intracellular complement and single-cell metabolism are closely connected at least in part because lysosomes and mitochondria, which are key components of cellular metabolic machinery53, are hotspots of complosome activity. For example, in several cell populations, the lysosome contains stores of C3, which are cleaved by the protease cathepsin L (CTSL) into C3a and C3b8. Lysosomes across immune cells also contain stores of C5 and C5a, and C5 activation — at least in monocytes and macrophages — seems to be dependent on an intracellular C5 convertase43. Intrinsic engagement of lysosomal C3aR promotes the low-level activation of mammalian target of rapamycin (mTOR) that generally supports cell survival54. In addition, cell-autonomous activation and processing of CD46 initiates the assembly of the mTOR complex 1 (mTORC1) at the lysosomal surface9,55. mTORC1 is a major nutrient-sensing node and controller of cellular growth that also regulates the cholesterol biosynthesis pathway via a currently undefined process56. CD46 induces the expression of genes that encode glucose transporters, such as SLC2A1 encoding glucose transporter 1 (GLUT1), to enable the nutrient influx required for cell activation. Collectively, these events drive the rise in glycolysis, fatty acid metabolism and oxidative phosphorylation that generally underlie the initiation of cell effector functions9,57,58. Moreover, C3aR and C5aR1 are expressed on, or can be translocated to, the mitochondrial outer membrane, where their stimulation controls Ca2+ flux47 and the direction of the electron transport chain43 via extracellular signalling kinase (ERK) 1/2-induced signals. Those signalling events control the production of reactive oxygen species versus ATP, and the net cellular induction of glycolysis and oxidative phosphorylation42,43. Lack of intracellular C5 or mitochondrial C5aR1 activity can affect the mitochondrial footprint (that is, the overall volume or space that mitochondria occupy in a cell), network morphology (that is, the overall balance of mitochondrial fission and fusion in a cell) and cell survival43, which suggests that the complosome contributes to maintenance of normal mitochondrial function and fitness59.
Autophagy and vesicular transport
The complosome is also involved in autophagy, which is intimately intertwined with cell metabolic processes60. Specifically, intracellular C3 or C3b participate in anti-bactericidal autophagy44,61, cell-protective autophagy in response to environmental stress11 and tonic lipophagy (a specialized form of autophagy62) for the maintenance of liver triglyceride levels63 (discussed in more detail below). Of note, thus far, C3-mediated control of autophagy seems to be driven by direct interaction of C3 fragments with autophagy related 16 like 1 (ATG16L1), independently of the activation of intracellular C3 fragment receptors.
Cell-specific activation (for example, T cell receptor engagement in T cells or TLR stimulation in myeloid cells) can trigger the activation and/or re-distribution of complosome components, including complement regulators and receptors, between sub-cellular compartments8,9,43,64. Interestingly, intracellular complement components seem to have an active role in this reshuffling of sub-compartmental content.
Gene expression and protein translation
Unexpectedly, the nucleus is another functional location of the complosome. For example, human CD46 can have a direct and active role in gene transcription and regulation. CD46 exists in isoforms with two different cytoplasmic signalling domains, CYT-1 and CYT-2. Following CD46 stimulation, CYT-1 and CYT-2 are released by the γ-secretase complex65 and translocate to the nucleus31. The nuclear translocation of CYT-1 and CYT-2 are crucial to the induction of metabolic changes driven by CD46 (ref. 31), which include the transcription of genes that encode nutrient channels such as SLC2A1 and SLC7A5 (which encodes large neutral amino acid transporter 1 (LAT1))9, as well as genes that encode metabolic enzymes58. Notably, CYT-1 and CYT-2 lack a DNA-binding site and thus cannot function directly as transcription factors; the exact mechanism underlying CD46-controlled gene activation remains to be defined66. Cleaved CYT-2 is also involved in protein translation. In bladder epithelial cells, CYT-2 binds to heterogeneous nuclear ribonucleoprotein (hnRNP) A1 directly, which facilitates mRNA translation through assembly of the translational machinery45. Finally, C3 has also been implicated in the regulation of histone–DNA interactions10.
The complosome across cells and tissues
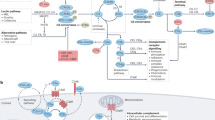
The non-canonical functions of intracellular complement components seem to support normal cell-specific behaviour, homeostasis and/or responses (Fig. 2). Moreover, although initially discovered in human CD4+ T cells8, the complosome has been detected across a range of cells and tissues, where it seems to function in a cell-specific manner (Fig. 2).

The figure illustrates the cells and tissues in which intracellular C3 or C5, or other complement components and receptors have been observed. The C3 and C5 activation fragments that drive the different cellular functions and biological pathways are not shown for simplicity and because they are not known in all cases. Cell-specific activities and overall contributions to normal tissue function of the complosome are shown to the right of each cell and tissue type. The blue arrow below indicates that tissue-resident immune cells (and their complosome) probably also participate in normal tissue homeostasis. C3aR, C3a receptor; C5aR1 or 2, C5a receptor 1 or 2; ETC, electron transport chain; FH, factor H; FHR-3, factor H-related protein 3; MAVS, mitochondrial anti-viral signalling protein; NF-κB, nuclear factor κB; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3 inflammasome; OXPHOS, oxidative phosphorylation; RIG-I, retinoic acid-inducible gene-I; ROS, reactive oxygen species; SLEC, short lived effector T cell; TH1, T helper 1; VLDL, very low density lipoprotein.
Immune cells
T cells
Intracellular C3 stores in human circulating CD4+ T cells not only provide a homeostatic survival signal via tonic mTOR activation but are also vital for the induction and contraction of TH1 effector responses. Antigen binding via the T cell receptor and CD28 engagement on human CD4+ T cells induces the rapid shuttling of intracellularly generated C3b to the T cell surface where it engages CD46 (ref. 8). Such cell-intrinsic CD46 stimulation triggers nuclear translocation of the cytoplasmic domains of CD46 and the metabolic reprogramming specifically required for IFNγ production and TH1 cell induction8,9,67 (Fig. 2). The CD46 cleavage triggered by T cell activation is temporally controlled and contributes to the TH1 cell shutdown program (via control of cholesterol efflux) and cessation of TH1 cell responses following pathogen clearance65,68,69. These effects of the cleaved intracellular CD46 domains converge with human T cell-autonomous generation of C5a that binds to mitochondrial C5aR1 and leads to assembly of the NOD-, LRR and pyrin domain-containing protein 3 inflammasome (NLRP3) assembly and IL-1β secretion42. This effect supports TH1 cell responses at mucosal interfaces42. Of note, the alternative C5a receptor (C5aR2) expressed on the surface of TH1 cells seems to inhibit NLRP3 activation and IL-1β production via a currently undefined mechanism, and also contributes to TH1 contraction42,70.
The complosome is also an integral part of cytolytic T cell (CTL) responses. In human CD8+ T cells, intrinsic CD46 engagement is required for the heightened glycolysis levels and fatty acid synthesis that underlie optimal IFNγ secretion, expression of granzyme B and cytotoxicity58,71. Furthermore, and similar to what is observed in CD4+ T cells, intracellular complement components can negatively regulate CTL responses. For example, the complement-derived pattern recognition molecule C1q, which is sourced from the extracellular environment, acts on human and mouse CD8+ T cell mitochondria to restrain glycolysis and effector function in the setting of infection to reduce pathological tissue conditions72.
B cells
Although non-activated human and mouse B cells contain stores of intracellular C3 and C3a8,73, whether cell-autonomous C3 generation is crucial to normal B cell survival and function is currently debated. However, one study showed that, in human B cells, internalized C3 and C3a can enter the nucleus and bind histones; this binding might control DNA packaging, as C3 inhibited histone–DNA interactions, but this notion needs to be further substantiated10. Of note, this study focused on the source of intracellular C3 and its subcellular distribution, but did not explore the potential effects of reduced C3 uptake on B cell functions such as cytokine production or antibody generation. Conversely, a separate study demonstrated the importance of extracellular C3 for normal human B cell memory formation but did not investigate whether this effect was dependent on C3 uptake and/or subsequent intracellular C3 activities74.
B cell-expressed anaphylatoxin receptors are emerging as important contributors to normal germinal centre formation28 and, although systemic C3 is the dominant source of the C3a that binds C3aR on B cells, some evidence indicates a less prominent but still significant effect of B cell-derived C3 in this process28. However, whether the effect of such B cell autonomous C3 production is rooted in intracellular C3 activity or in the activity of secreted C3 that functions in an autocrine or paracrine fashion remains unclear. Overall, complosome contributions to normal B cell biology remain largely unmapped, but might be an interesting area for further investigation.
Neutrophils, monocytes and macrophages
Although most immune cells in circulation are characterized by relatively low C3 and C5 expression, blood-derived neutrophils and classic monocytes continuously transcribe high levels of C3 (ref. 75) and C5 (ref. 43) in mice and humans. Interestingly, neutrophils and classic monocytes are also frontline immune cell sentinels during the detection of noxious microbes and their exceptionally quick cellular responses are essential to effective pathogen elimination76. Whether sustained high C3 loading in neutrophils and classic monocytes is required for their ‘rapid response program’ remains to be investigated. Of note, one study suggested that the neutrophil complosome might control cellular biomechanics via cytoskeletal re-arrangement77. The exact molecular mechanism has not been defined, but complosome-controlled cytoskeletal activity in neutrophils reduced the stiffness of the cell membrane and supported neutrophil movement through small vessels.
Macrophages and monocytes rely on cell-autonomous and intracellular C3 and C5 activation — specifically, the engagement of mitochondrial C5aR1 — for optimal metabolic reprogramming and NLRP3 inflammasome activation to produce normal levels of pro-inflammatory IL-1β during protective immunity and sterile inflammation43,75. Conversely, C1q bound to apoptotic cells and therefore ingested by human macrophages as they phagocytose these cells, inhibits NLRP3 activity and thus limits their polarization towards an M1 phenotype. This regulatory effect promotes the non-inflammatory removal of dead cells78.
Intracellular complement also controls another key aspect of macrophage biology — the processing of antigens. Through a molecular process that has not yet been defined, intracellular C3b-opsonization of antigens ingested by mouse macrophages and transported into the lysosome, slows the fusion of cargo-laden lysosomes with phagosomes79. This C3b-mediated effect ‘paces’ antigen processing and subsequent macrophage-directed T cell responses79. Surprisingly, intracellular complement activity can also actively control the opsonization of target cells. FH is internalized by early apoptotic human cells and supports CTSL-mediated cell-intrinsic C3 processing and surface iC3b deposition to support safe clearance of the dying cell by phagocytes, as iC3b recognition by its receptor (CR3; comprising CD18 and CD11b) does not lead to cell activation80. Moreover, akin to what is seen in T cells, the complosome also contributes to human and mouse myeloid cell homeostasis because tonic mitochondrial C5aR1 signalling sustains the monocyte mitochondrial footprint, morphology and basic metabolic turn-over in the steady state43.
Non-immune cells and tissues
Intracellular complement activities extend beyond immune cells and have been observed in cells at important host–environment borders, tissue structural mesenchymal cells and highly specialized cell populations across tissues and organs (Fig. 2).
Epithelial and endothelial cells
Epithelial cells, which are crucial to interactions between the host and the external environment, can harbour or acquire a substantial range of complement components. The first observations of intracellular C3 activity in epithelial cells were made during studies that focused on the barrier function of epithelial cells during infections. For example, the C3b that opsonizes Listeria monocytogenes bacteria and enters the human epithelial cell cytoplasm during bacterial invasion supports autophagy-mediated bacterial clearance via a direct interaction with ATGL16L1 (ref. 44). Moreover, C3b-opsonized viruses trigger human epithelial cell-autonomous immunity during infection through activation of the mitochondrial antiviral signalling (MAVS) pathway, which restricts virus replication81. C1qA within epithelial cells can also interact directly with components of the retinoic acid-inducible gene-I (RIG-I) pathway and enhance anti-viral responses via nuclear factor-κB (NF-κB)-driven IFNβ production82 (Box 2). Notably, pathogens can also take advantage of complosome components to promote infection. For example, CD46 serves as a cell entry receptor for several important human pathogens and signalling events induced by pathogen binding to CD46 on epithelial cells support the invasion of bacteria and viruses across epithelial barriers83.
Importantly, the complosome also orchestrates epithelial cell responses towards non-infectious stressors. C3-regulatory factor H-related protein 3 (FHR-3), which is taken up by human retinal epithelial cells in the eye in conditions of oxidative stress, enhances intracellular complement activation and secretion of pro-inflammatory cytokines84, whereas mitochondrial C3aR stimulation within retinal epithelial cells reduces mitochondrial respiration and combats oxidative stress47. In human lung, intracellular C3 protects airway epithelial cells from nutrient deprivation-induced cell death via a currently unknown mechanism85. C5 expression has also been observed in healthy human and mouse type II alveolar epithelial cells; however, its functional relevance has not yet been explored86. In bladder epithelial cells, the processed CYT-2 domain of CD46 controls hypoxia-inducible factor 1α (HIF1α) and is associated with malignant transformation and bladder cancer, probably via hyperactivation of MYC-mediated cell proliferation45. Similarly, intracellular C1q binding protein (C1QBP) sustains normal division cycles in human intestinal epithelial cells by controlling mitochondrial activity87 (Fig. 2).
Our insights into the diverse roles of the complosome in the epithelium are broadening but our knowledge about the complosome in endothelial cell biology is sparse. A single study exploring the role of the complosome in kidney glomerular endothelial cells indicates that it contributes to normal endothelial cell homeostasis because the absence of cell-intrinsic intracellular FH caused spontaneous cytoskeletal remodelling, aberrant cellular layer integrity with changes in cell morphology and loss of barrier function, increased cell proliferation owing to changes in mitochondrial respiration and ATP production, and overall heightened angiogenic potential in human and mouse kidney endothelial cells88. Functionally, intracellular FH seems to repress inflammation-associated gene programmes by restricting the nuclear translocation of NF-κB in these cells but the underlying mechanism was not identified. Understanding the role of the complosome in vascular endothelial cells will be highly valuable as these cells might represent an important interface for cross-communication between the complosome and the classic circulating complement system in health and disease.
Fibroblasts and mesothelial cells
Fibroblasts are key mesenchymal cells that contribute to the structural framework and volume of tissues89. An exciting study found that induction of C3 expression in fibroblasts of mouse arthritic joints contributes to local immune priming and the exacerbation of immune responses upon antigen rechallenge or re-encounter90. Specifically, an initial antigenic insult in the joint augmented C3 expression in synovial fibroblasts and induced a state of priming. This effect relied on the activation of intracellular complement C3 and C3a receptors, and downstream mTOR and HIF1α-controlled metabolic reprogramming, which ultimately enhanced NLRP3 activity and sensitized the tissue to inflammation. This study focused on synovial fibroblasts, but the complosome might also have a role in fibroblasts in other tissues.
Interestingly, a 2022 study that mapped complosome expression across all cell populations of healthy mouse and human lungs noted that mesothelial cells expressed the greatest number of complement genes (core components and regulators)86. Mesothelial cells form the protective lining of serous body cavities91,92, where they mediate the transport of fluids and cells across organ linings, sample the microenvironment through phagocytosis and produce cytokines92. Importantly, mesothelial cells have stem cell-like characteristics and can give rise to fibroblasts during the initiation of tissue repair93; both cell types are key participants in tissue homeostasis, inflammation and repair89. These cells also represent essential nodes in stromal–immune crosstalk, and therefore the presence of the complosome in these cells might have important implications for intracellular complement activities across tissues and organs, both in healthy tissue biology and fibroblast-associated human diseases.
Hepatocytes
In addition to key structural or functional constituents of all major organs, such as fibroblasts and mesothelial cells, the complosome is present in highly specialized and tissue-restricted non-immune cells such as hepatocytes. Importantly, hepatocytes and the liver are the principal source of circulating complement94 and C3 contributes to normal liver function and regeneration following injury or exposure to toxins95. Although the effects of C3 on the liver were thought to be solely mediated by extracellular C3, emerging evidence indicates that the complosome also contributes to normal hepatocyte function. Specifically, intracellular C3 in mouse hepatocytes prevents hepatic steatosis by controlling basal liver triglyceride levels through sustained low-level lipophagy, which maintains normal secretion of very-low-density lipoprotein63. Given the central role of the liver in complement biology, it will be worth assessing whether other complosome components are intracellularly active in hepatocytes and whether they control additional cell physiological processes.
Neurons
The observation that complement in the brain contributes to normal development of the central nervous system, mediates nerve pruning (that is, the removal of unwanted neuronal connections during neuronal network formation)96,97,98, can affect memory and behaviour96,99,100, and contributes to several pathological neurological conditions, such as Alzheimer’s disease101,102, Parkinson’s disease103 and schizophrenia104, renewed interest in the complement system among neuro-immunologists. However, the source of complement components in the brain is not well delineated and current hypotheses include influx from the circulation via a compromised blood–brain barrier105 or expression by brain cells106. Whether intracellularly active complement components are present in the healthy human brain and contribute to normal brain and central nervous system development and/or function remains unclear, but C1q has been observed within neurons of the rhesus macaque, although its functional consequences have not been explored107. In mice, C1q within cortical neurons can activate mitochondria, which leads to reactive oxygen species production, changes in mitochondrial respiration and augmented neuronal activity108.
Pancreatic β-cells
The complosome is also active in endocrine cells and is crucial to the function of pancreatic β-cells. The glycosylphosphatidylinositol (GPI)-linked complement regulator CD59 is normally expressed on cell surfaces to protect host cells from MAC attack and lysis109; however, human and mouse pancreatic β-cells express an exclusively intracellular non-GPI-anchored form of CD59110,111. Within β-cells, this CD59 form interacts with the cytosolic exocytosis machinery that mediates the secretion of insulin in response to raised circulating glucose levels112, including the exocytotic SNARE proteins vesicle-associated membrane protein 2 (VAMP2) and synaptosome-associated protein 25 (SNAP25). Intracellular C3 is also an important complosome component in pancreatic β-cells, where it induces protective autophagy via interaction with ATGL16L1 during glycolytic stress, which in turn maintains β-cell survival and thus normal glucose levels and metabolism in the host113.
The complosome in human disease
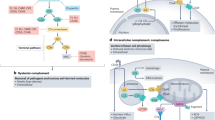
Complement dysregulation is associated with many human diseases as members of the complement system can drive pathological conditions directly and/or exacerbate damage induced by non-complement disease initiators. In addition to the known role of systemic and extracellular complement in human disease (as previously reviewed23,24,114,115,116), here we focus on the contributions of intracellular complement (Fig. 3).

The figure shows disease states that are associated with pathological alterations in the expression levels or activity of specific intracellular complement components. Included in the lower right rectangle are additional pathological conditions in which complosome dysregulation might have a role, but for which solid evidence is still missing. AD, Alzheimer’s disease; AMD, age-related macular degeneration; C1qBP, C1q binding protein (also known as globular head C1q receptor, gC1qR); C5aR1, C5a receptor 1; COPD, chronic obstructive pulmonary disease; FH, factor H; FHR-3, factor H-related protein 3; IBD, inflammatory bowel disease; MS, multiple sclerosis; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; SLE, systemic lupus erythematosus.
Infections and infection-associated pathological conditions of tissue
Reductions in cell-autonomous C3 and/or C5 generation, or CD46 expression in human CD4+ and CD8+ T cells are associated with failure to induce protective TH1 cell and CTL responses, respectively8,31,42,58. Affected individuals have recurrent respiratory tract infections, at least early in childhood8,31,117,118 and, in rare cases, can develop common variable immune deficiency31. As discussed earlier, defects in intracellular complement activity in B cells and its potential impact on antibody responses in infection have not yet been explored. However, the inability of human and mouse monocytes and macrophages to increase LFA-1-driven C3 expression and intracellular C3 production upon diapedesis impairs their pro-inflammatory cytokine production program (with a specific defect in IL-1β production)75: patients with LFA-1 deficiency, which is a disorder termed leukocyte adhesion deficiency-1 (LAD-1), cannot boost C3 levels in immune cells, and this defect is key to the primary immune deficiency observed in these patients75.The reduction of IL-1β production by macrophages from LAD-1 patients could in part be rescued by adenovirus-mediated delivery of intracellular C3a, suggesting a contribution of intracellular C3 to normal cell activity75. Unfortunately, uncontrolled complosome activity can also promote the pathological conditions of tissue that can accompany most protective immune responses. For example, SARS-CoV-2-induced cell-intrinsic C3 and C3a generation in the human lung epithelium drives the hyper-activation of local complement that is associated with the development of severe COVID-19 (refs. 119,120,121,122).
Arthritic diseases
Unwanted or chronic activation of the complosome has emerged as a novel and central driver of arthritic disease. For example, sustained augmented C3 expression or perturbations in cell-intrinsic CD46-mediated signalling contribute to the destructive hyper-TH1 cell responses observed in the joints of patients with rheumatoid arthritis68,75. In fact, we identified increased C3 expression in T cells as a novel, highly specific, biomarker of rheumatoid arthritis severity75. This finding aligns with the observation that C3-mediated priming of synovial fibroblasts contributes to inflammation and tissue destruction in the joints of patients with rheumatoid arthritis90. Accordingly, a 2022 study that comprehensively assessed the expression of complement genes across cells of inflamed tissue from patients with rheumatoid arthritis revealed substantial regional alterations in activating and inhibitory factors that probably promote local complement activation123. Although the researchers did not examine the contribution of extra versus intracellular complement, the work provides an important and needed starting point for such future investigations.
In addition to changes in C3 and CD46, patients with systemic sclerosis123 and/or systemic lupus erythematosus124 have increased the activity of cell-intrinsic C5 and C5aR1 in injury-causing TH1 cells and CTLs compared with healthy controls. Moreover, lack of FH, which helps in the tagging of apoptotic cells with iC3b for removal by phagocytes, has been related to the induction of systemic lupus erythematosus as the absence of FH increased disease severity in lupus-prone mice80. Finally, increased production and cleavage activation of intracellular C3 and C5 in neutrophils were associated with the increased activity of low-density granulocytes, which are a distinct subset of highly vasculopathic neutrophils that is expanded in systemic lupus erythematosus77.
Atherosclerosis
Overactive mitochondrial C5aR1 activity and increased levels of cell-intrinsic C5 in monocytes and macrophages have been identified as important contributors to cardiovascular disease as they promoted the generation of IL-1β-producing M1-like macrophages in the aortic root of a mouse model of high fat diet-induced atherosclerosis, and in the arterial plaques of patients with cardiovascular disease43. Mechanistically, the accumulation of cholesterol induced by a high-fat diet triggered the continuous hyperactivation of mitochondrial C5aR1 via augmented intracellular C5a generation in monocytes and macrophages, which led to a change in the direction of the electron transport chain that increased glycolysis, promoted maladaptive IL-1β production and sustained an M1 phenotype43. Excitingly, a 2022 study identified C5 polymorphisms as a top risk factor for cardiovascular disease in humans125, although future work will need to determine whether intracellular and/or extracellular C5 drive this risk.
Diabetes and metabolic syndrome
Similar to atherosclerosis, studies on a potential pathological role of the complosome in diabetes and/or metabolic syndrome are limited. Nonetheless, the few data available — including the aforementioned roles of CD59 and C3 in pancreatic β-cells and insulin production11,111, and of triglyceride regulation via C3 expression in hepatocytes63 — are exciting and warrant further research. In addition, islet cells from patients with type 2 diabetes express significantly less insulin-controlling CD59 isoform than islet cells from healthy donors110.
Inflammatory bowel disease
As discussed earlier, the complosome is highly active in epithelial cells at mucosal sites (Fig. 2) and, unsurprisingly, deviations in normal complosome function in these cells might contribute to inflammatory bowel disease. For example, TLR-induced increased expression of colonic cell-intrinsic C3 induced a C3aR-dependent pro-inflammatory state in in vitro and in vivo LPS-stimulated mouse colon epithelial cells126. Pathological upregulation of C3 was also noted in a mouse model of dextran sulfate sodium-induced colitis, and C3 was elevated in the colonic epithelia of patients with Crohn’s disease but not of those with ulcerative colitis126. These data align with those of another study showing that intestinal epithelial cell intracellular C3 and C3a contribute to the tissue damage caused by hypoxia during ischaemic injury127. The exact molecular mechanism of action exerted by C3 and C3aR in the gut epithelium requires further investigation.
Cancer
The role of classic and local complement in cancer is under intensive investigation (as previously reviewed128,129) but the potential contributions of the complosome to malignancies remain largely unexplored. Nonetheless, some studies have suggested a role for the complosome in cancer induction and progression. For example, intestinal epithelial cells from patients with colorectal carcinoma had significantly reduced intracellular levels of globular C1qR (gC1qR) compared with colon cells from healthy individuals87. In addition, gC1qR1, which is located largely at the mitochondria in healthy colon cells and supports oxidative phosphorylation, remained in a non-mitochondrial intracellular location in patient cells; this alternative localization was associated with augmented glycolysis, cell proliferation and intestinal malignant transformation87. Further, tumour grading correlated significantly with reduced localization of gC1qR to mitochondria in patients with colorectal carcinoma87. Although normal human lung epithelial cells seem to be devoid of basal FH expression, in patients with lung adenocarcinoma, intracellular expression of FH was surprisingly observed in tumour cells and this aberrant FH presence was associated with decreased control of normal cell cycling and with pro-tumoural transformation130.
FH is also overexpressed in human clear-cell renal cell carcinoma (ccRCC)130 and not only is the structure of intracellular FH in ccRCC similar to that of FH secreted by the liver, this protein can also function as a C3 regulator. Although the exact mechanism has not yet been defined, FH seems to support malignant transformation in ccRCC in a process that involves either direct or indirect regulation of the transcriptome. Silencing of FH in ccRCC cells but not in healthy kidney tubular cells led to significant changes in the expression of numerous genes involved in the control of cell proliferation130. Of note, this pro-tumour effect of FH was independent of intracellular C3 or C5 activation.
Intracellular C1 molecules might also have an unconventional and complement cascade-independent role in ccRCC tumour proliferation and survival131. High cell-intrinsic C1 expression in tumours is associated with poor prognosis and seems to support a tumorigenic environment by supporting not only the proliferation of the tumour cells but also the influx of T cells and macrophages with a more suppressive phenotype into the tumour vicinity131. This observation suggests a role for the complosome in the communication between kidney parenchymal and resident and/or infiltrating immune cells, an exciting and important prospect that should be explored in the future.
Kidney disease
The kidney seems particularly prone to the accumulation of activated complement fragment deposits in diseases where systemic or local complement activation is observed14,132. The exact molecular reasons underlying this deposition remain unclear, but high hydrostatic pressure in combination with fluid absorption and filtration functions in the kidney might increase its susceptibility to changes in the expression of complement inhibitors133. Moreover, C3 derived from kidney tubular epithelial cells is a crucial contributor to graft rejection in kidney transplantation134,135. Accumulating evidence suggests that changes in the intracellular activities of complement components in kidney endothelial or epithelial cells contribute to organ dysfunction in the steady state88 and fibrosis after kidney injury43.
As discussed earlier, the tonic presence of intracellular FH in kidney glomerular endothelial cells is a pre-requisite for their normal function in the resting state88. Of note, the researchers did not investigate in detail whether cell-intrinsic intracellular FH is similar in structure and function to circulating FH, nor did they conclusively exclude the possibility that the intercellular activity of FH partly operates via regulation of intracellular C3. In contrast to kidney endothelial cells, intracellular FH is normally not detectable in tubular epithelial cells in the steady state and, accordingly, CFH knockdown had no effect on their transcriptional programme or cellular behaviour130.
In a folic acid-induced tubular cell injury model, intracellular C5 and C5aR1 activity in kidney-infiltrating macrophages seems to contribute to the observed nephropathy. Specifically, mice with macrophages deficient for C5 or C5aR1 had reduced kidney fibrosis compared with wild type controls and mitochondrial C5aR1 hyper-activation was identified as a driver of the initial folic acid-induced injury43,136. Furthermore, single-cell RNA sequencing of kidney tissue showed that unilateral ureteral obstruction in mice increases kidney C3 and C5 expression, mostly in tubular epithelial cells, which is accompanied by a rise in C3aR and C5aR1 expression in kidney interstitial immune cells136. We anticipate that aberrant complosome activity might also contribute to other pathological kidney conditions, such as C3 glomerulopathies and lupus nephritis (Fig. 3). Assessing whether intracellular complement contributes to kidney fibrosis will also be of particular interest given its central role in the development of kidney failure137.
Therapeutic implications
Complement perturbations, including deviations in normal complosome activity, can cause several prevalent human diseases, as discussed above. However, success in developing broad and effective complement therapeutics has been modest — currently, ten drugs are available in the clinic and their application is limited to the treatment of rare conditions such as angioedema, haemolytic uraemic syndrome or paroxysmal nocturnal haemoglobinuria114. Importantly, these traditional therapeutics only target serum-operative or extracellularly active complement. Given the emerging prominent role for the complosome in the control of basic cell physiology across immune and non-immune cells in health and disease, tackling intracellular complement, possibly in combination with the traditional extracellular complement, might provide a novel approach towards more broadly effective complement therapeutics. Of note, improved patient stratification within studies would also help to elucidate complosome perturbations, and aid therapeutic development. Here, defining the patient complotype (that is, the combination of polymorphisms across complement genes138) might be especially useful.
Application of a cell-permeable CTSL inhibitor normalized the pathologically high production of IFNγ by TH1 cells isolated from the inflamed joint of patients with idiopathic juvenile arthritis in vitro to normal levels seen in paired TH1 cells from the blood of these patients8 and a cell-permeable FB inhibitor abrogated SARS-CoV2-triggered intracellular hyper-C3 activation in human lung epithelial cells in vitro119. The same cell-permeable FB inhibitor also reduced macrophage hyperactive responses in isolated unstable arterial plaques of patients with cardiovascular disease compared with carrier-treated isolated arterial plaques43. Finally, we determined that the peptide-based C5aR1-antagonist JPE1375 is cell-permeable43 and can successfully ablate mitochondrial C5aR1 signalling, which reduced pro-inflammatory and atherosclerosis-driving IL-1β production in cholesterol-crystal sensing human monocytes in vitro. Given these data, assessing other, existing, small-molecule complement inhibitors for their ability to pass cellular membranes and their potential effects on the complosome will also be of interest.
Targeting upstream inducers of complosome expression and activity is another emerging strategy. For example, cholesterol crystals are known complosome activators139 and, in a murine atherosclerosis model, treatment with oligosaccharide 2-hydroxypropyl-β-cyclodextrin, which is a compound that increases cholesterol solubility and can reverse atherosclerosis, reduced inflammation-driven complosome component expression in plaque cells140,141. Moreover, type I interferons control C3 expression during viral infections and Janus kinase (JAK)1/2 inhibitors, which ablate type I interferon receptor signalling, have thus been proposed as a potential approach to reducing complosome activity119. Finally, the integrin lymphocyte function-associated antigen 1 (LFA-1) has been identified as a master regulator of C3 expression in immune cells75,142 and might therefore also warrant consideration as a cell-surface target for intracellular C3 control.
Importantly, several currently existing limitations and caveats might hamper the successful development of complosome-targeted drugs. For example, the molecular mechanisms underlying complosome activities, how they are regulated, and how they intersect not only with other intracellular pathway systems but also with the extracellular, canonical complement system must be clarified. However, the current difficulties in dissecting conclusively, and specifically in vivo, which parts of cell-associated complement originated in a cell-intrinsic versus cell-extrinsic manner, and which components function within the cell or on the cell surface (upon secretion) hinder this goal. For example, currently no methods enable the specific ablation of sub-cellular but not surface complement receptor expression. Generating deeper insights into complosome biology should ideally be accompanied by the improvement of small animal and/or preclinical models, including the generation of mice with cell-specific and inducible ablation of complosome components. However, important species-specific differences in complosome biology exist between mice and man86 and these must be taken into consideration. For example, rodents do not express CD46 on somatic tissues and a functional murine homologue that serves the diverse roles of CD46 has not yet been identified143. In addition, the expression and activity of C3aR and C5aRs by mouse T cells is still disputed in the field17. Of note, data supporting our early suggestion that cell-autonomous C3 differs in structure and post-translational modifications from liver-derived C3 (and might therefore not be recognized by antibodies raised against the latter C3 form144) is only now emerging145. Such differences will probably require a new range of highly specific reagents to be developed to probe complosome biology that take into account its unique biochemistry (reviewed in ref. 64).
Specific details on how the normal complosome changes in different human diseases, and its functional consequences on disease pathology and/or progression must also be clarified. These insights are especially important where the same complosome components might contribute to pathological conditions, while also being essential for tissue homeostasis in the steady state and/or repair and return to homeostasis following an immune response. Furthermore, complosome activity occurs across cellular systems during an immune response, often in a stimulus-specific (and temporal) fashion. Specific targeting of complosome functions and cell types (and possibly subcellular compartments) might therefore be required to avoid undesired side effects.
Conclusions
Current evidence suggests that the complement system is ubiquitously present in the host — from subcellular compartments, the cytoplasm and cell surfaces to the extracellular space, and the interstitial and body fluids. Furthermore, the location of complement activity seems to dictate its diverse functions, with circulating complement protecting the vascular space from bloodborne infections, immune cell-secreted and locally active complement directing immune cell effector function, and intracellular complement orchestrating basic cell physiology.
Intracellular complement activities have been observed in all cell types assessed so far for its presence, indicating its broad physiological importance. Accordingly, the complosome contributes to normal cell turnover in the steady state, cell-specific responses to infectious and non-infectious stressors, and the subsequent return to homeostasis. We suggest that the close connection between the complosome and cellular physiology machinery might be rooted in the evolutionary origin of C3 as a single-cell intracellular protein with non-canonical metabolic functions50. The liver-derived and secreted version, with its known canonical activities, might have appeared later during the evolution of multi-organ organisms that required protection of the vasculature. Here, we have detailed the currently known complosome contributions within distinct cell populations in isolation. We fully expect that immune cell subtypes not yet probed for intracellular complement presence — such as natural killer cells, natural killer T cells, dendritic cells, γδ T cells, mast cells and eosinophils — and other non-immune cells will join this list in the future. Understanding how the complosome might contribute to communication between cells, for example, during immune cell influx into tissues upon infection and inflammation will also be important (Fig. 2).
Although the complosome has been detected in several major organs, whether complosome dysfunction always contributes to diseases associated with these sites remains unclear. In this regard, we suggest that, in addition to the diseases discussed earlier, the role of the complosome in age-related macular degeneration or general pathological conditions of tissue associated with ageing84, additional pulmonary disease conditions (such as chronic obstructive pulmonary disease and asthma)86,146, chronic inflammatory conditions of the brain147, and possibly diseases associated with faulty haemopoiesis148 should be investigated further (Fig. 3). Moreover, as a community, we may need to reassess patients and our clinical data, as well as reapply and reinterpret animal models of complement-driven pathophysiologies for missed contributions of the complosome.
Detangling the specific individual and interrelated contributions of extracellular and intracellular complement to host homeostasis, as well as their respective functional intersections with other extracellular and intracellular biological effector systems is now essential. The novel concept of the complosome is receiving increasing attention among the broader immunological community and underpins the notion that complement is by no means fully explored. These insights have been fuelled in part by the improved general accessibility of key technologies, including single-cell RNA sequencing and proteomics, and the observation that central complement components often rank prominently among candidate genes and pathways that differ between healthy and diseased cells and tissues.
The observation that complement ‘is everywhere’ and, unexpectedly, controls fundamental cellular processes from within cells probably explains why so many human diseases are associated with complement perturbations. Given that the complosome largely functions beyond the classically known complement activities, dissecting its biology will require tissue-focused, cutting-edge technological approaches, as well as collaborative efforts among scientists and clinicians across a diverse range of disciplines. We argue that the collective new knowledge gained by such approaches will be of great value for our understanding of complement biology in general and its translation into future therapeutic benefit.
References
Bordet, J. G. O. Sur l’existence de substances sensibilisatrices dans la plupart des serums antimicrobiens. Ann. De. l’Inst. Pasteur 15, 289–303 (1901).
Merle, N. S., Church, S. E., Fremeaux-Bacchi, V. & Roumenina, L. T. Complement system part I — molecular mechanisms of activation and regulation. Front. Immunol. 6, 262 (2015).
Merle, N. S., Noe, R., Halbwachs-Mecarelli, L., Fremeaux-Bacchi, V. & Roumenina, L. T. Complement system part II: role in immunity. Front. Immunol. 6, 257 (2015).
Pepys, M. B. Role of complement in induction of the allergic response. Nat. N. Biol. 237, 157–159 (1972).
Killick, J., Morisse, G., Sieger, D. & Astier, A. L. Complement as a regulator of adaptive immunity. Semin. Immunopathol. 40, 37–48 (2018).
Carroll, M. C. & Isenman, D. E. Regulation of humoral immunity by complement. Immunity 37, 199–207 (2012).
Kolev, M., Le Friec, G. & Kemper, C. Complement — tapping into new sites and effector systems. Nat. Rev. Immunol. 14, 811–820 (2014).
Liszewski, M. K. et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 39, 1143–1157 (2013).
Kolev, M. et al. Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity 42, 1033–1047 (2015).
Kremlitzka, M. et al. Interaction of serum-derived and internalized C3 with DNA in human B cells — a potential involvement in regulation of gene transcription. Front. Immunol. 10, 493 (2019).
King, B. C., Renstrom, E. & Blom, A. M. Intracellular cytosolic complement component C3 regulates cytoprotective autophagy in pancreatic β cells by interaction with ATG16L1. Autophagy 15, 919–921 (2019).
West, E. E., Kolev, M. & Kemper, C. Complement and the regulation of T cell responses. Annu. Rev. Immunol. 36, 309–338 (2018).
Kolev, M. et al. Inside-out of complement in cancer. Front. Immunol. 13, 931273 (2022).
Freiwald, T. & Afzali, B. Renal diseases and the role of complement: linking complement to immune effector pathways and therapeutics. Adv. Immunol. 152, 1–81 (2021).
Hajishengallis, G., Reis, E. S., Mastellos, D. C., Ricklin, D. & Lambris, J. D. Novel mechanisms and functions of complement. Nat. Immunol. 18, 1288–1298 (2017).
Reis, E. S., Mastellos, D. C., Hajishengallis, G. & Lambris, J. D. New insights into the immune functions of complement. Nat. Rev. Immunol. 19, 503–516 (2019).
Laumonnier, Y., Karsten, C. M. & Kohl, J. Novel insights into the expression pattern of anaphylatoxin receptors in mice and men. Mol. Immunol. 89, 44–58 (2017).
Bosmann, M. & Ward, P. A. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv. Exp. Med. Biol. 946, 147–159 (2012).
Mathern, D. R. & Heeger, P. S. Molecules great and small: the complement system. Clin. J. Am. Soc. Nephrol. 10, 1636–1650 (2015).
West, E. E. & Kemper, C. in Paul’s Fundamental Immunology, 8th edn (eds Flajnik, M. F., Singh, N. J. & Holland, S. M.) 1–39 (Wolters Kluwer, 2022).
Fernandez-Sola, J. et al. Persistent low C3 levels associated with meningococcal meningitis and membranoproliferative glomerulonephritis. Am. J. Nephrol. 10, 426–430 (1990).
Defendi, F., Thielens, N. M., Clavarino, G., Cesbron, J. Y. & Dumestre-Perard, C. The immunopathology of complement proteins and innate immunity in autoimmune disease. Clin. Rev. Allergy Immunol. 58, 229–251 (2020).
Conigliaro, P. et al. Complement, infection, and autoimmunity. Curr. Opin. Rheumatol. 31, 532–541 (2019).
Pouw, R. B. & Ricklin, D. Tipping the balance: intricate roles of the complement system in disease and therapy. Semin. Immunopathol. 43, 757–771 (2021).
Liszewski, M. K. & Atkinson, J. P. Complement regulators in human disease: lessons from modern genetics. J. Intern. Med. 277, 294–305 (2015).
Cserhalmi, M., Papp, A., Brandus, B., Uzonyi, B. & Jozsi, M. Regulation of regulators: role of the complement factor H-related proteins. Semin. Immunol. 45, 101341 (2019).
Matsumoto, A. K. et al. Intersection of the complement and immune systems: a signal transduction complex of the B lymphocyte-containing complement receptor type 2 and CD19. J. Exp. Med. 173, 55–64 (1991).
Cumpelik, A. et al. Dynamic regulation of B cell complement signaling is integral to germinal center responses. Nat. Immunol. 22, 757–768 (2021).
Astier, A., Trescol-Biemont, M. C., Azocar, O., Lamouille, B. & Rabourdin-Combe, C. Cutting edge: CD46, a new costimulatory molecule for T cells, that induces p120CBL and LAT phosphorylation. J. Immunol. 164, 6091–6095 (2000).
Zaffran, Y. et al. CD46/CD3 costimulation induces morphological changes of human T cells and activation of Vav, Rac, and extracellular signal-regulated kinase mitogen-activated protein kinase. J. Immunol. 167, 6780–6785 (2001).
Le Friec, G. et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat. Immunol. 13, 1213–1221 (2012).
Heesterbeek, D. A. C., Angelier, M. L., Harrison, R. A. & Rooijakkers, S. H. M. Complement and bacterial infections: from molecular mechanisms to therapeutic applications. J. Innate Immun. 10, 455–464 (2018).
Botto, M. et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 19, 56–59 (1998).
Strainic, M. G. et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 28, 425–435 (2008).
Lalli, P. N. et al. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood 112, 1759–1766 (2008).
Lalli, P. N., Strainic, M. G., Lin, F., Medof, M. E. & Heeger, P. S. Decay accelerating factor can control T cell differentiation into IFN-γ-producing effector cells via regulating local C5a-induced IL-12 production. J. Immunol. 179, 5793–5802 (2007).
Fang, C., Miwa, T. & Song, W. C. Decay-accelerating factor regulates T-cell immunity in the context of inflammation by influencing costimulatory molecule expression on antigen-presenting cells. Blood 118, 1008–1014 (2011).
Liu, J. et al. IFN-γ and IL-17 production in experimental autoimmune encephalomyelitis depends on local APC-T cell complement production. J. Immunol. 180, 5882–5889 (2008).
Cravedi, P. et al. Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am. J. Transpl. 13, 2530–2539 (2013).
Pratt, J. R., Basheer, S. A. & Sacks, S. H. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat. Med. 8, 582–587 (2002).
Angeletti, A. et al. Loss of decay-accelerating factor triggers podocyte injury and glomerulosclerosis. J. Exp. Med. https://doi.org/10.1084/jem.20191699 (2020).
Arbore, G. et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science 352, aad1210 (2016).
Niyonzima, N. et al. Mitochondrial C5aR1 activity in macrophages controls IL-1β production underlying sterile inflammation. Sci. Immunol. 6, eabf2489 (2021).
Sorbara, M. T. et al. Complement C3 drives autophagy-dependent restriction of cyto-invasive bacteria. Cell Host Microbe 23, 644–652 e645 (2018).
Zeng, J. et al. CD46 splice variant enhances translation of specific mRNAs linked to an aggressive tumor cell phenotype in bladder cancer. Mol. Ther. Nucleic Acids 24, 140–153 (2021).
Ding, P. et al. Intracellular complement C5a/C5aR1 stabilizes β-catenin to promote colorectal tumorigenesis. Cell Rep. 39, 110851 (2022).
Ishii, M., Beeson, G., Beeson, C. & Rohrer, B. Mitochondrial C3a receptor activation in oxidatively stressed epithelial cells reduces mitochondrial respiration and metabolism. Front. Immunol. 12, 628062 (2021).
Karsten, C. M., Laumonnier, Y. & Kohl, J. Functional analysis of C5a effector responses in vitro and in vivo. Methods Mol. Biol. 1100, 291–304 (2014).
Hess, C. & Kemper, C. Complement-mediated regulation of metabolism and basic cellular processes. Immunity 45, 240–254 (2016).
Kolev, M. & Kemper, C. Keeping it all going — complement meets metabolism. Front. Immunol. 8, 1 (2017).
Liszewski, M. K., Elvington, M., Kulkarni, H. S. & Atkinson, J. P. Complement’s hidden arsenal: new insights and novel functions inside the cell. Mol. Immunol. 84, 2–9 (2017).
King, B. C. & Blom, A. M. Complement in metabolic disease: metaflammation and a two-edged sword. Semin. Immunopathol. 43, 829–841 (2021).
Wong, Y. C., Kim, S., Peng, W. & Krainc, D. Regulation and function of mitochondria-lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol. 29, 500–513 (2019).
Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 (2012).
Condon, K. J. & Sabatini, D. M. Nutrient regulation of mTORC1 at a glance. J. Cell Sci. https://doi.org/10.1242/jcs.222570 (2019).
Perucha, E. et al. The cholesterol biosynthesis pathway regulates IL-10 expression in human Th1 cells. Nat. Commun. 10, 498 (2019).
Xu, K. et al. Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Science 371, 405–410 (2021).
Arbore, G. et al. Complement receptor CD46 co-stimulates optimal human CD8+ T cell effector function via fatty acid metabolism. Nat. Commun. 9, 4186 (2018).
Rahman, J., Singh, P., Merle, N. S., Niyonzima, N. & Kemper, C. Complement’s favourite organelle — mitochondria? Br. J. Pharmacol. 178, 2771–2785 (2021).
Kim, K. H. & Lee, M. S. Autophagy — a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 10, 322–337 (2014).
Viret, C. et al. Regulation of anti-microbial autophagy by factors of the complement system. Microb. Cell 7, 93–105 (2020).
Zhang, S. et al. The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis. 13, 132 (2022).
Li, Y. et al. Intracellular C3 prevents hepatic steatosis by promoting autophagy and very-low-density lipoprotein secretion. FASEB J. 35, e22037 (2021).
King, B. C. & Blom, A. M. Intracellular complement: evidence, definitions, controversies, and solutions. Immunol. Rev. https://doi.org/10.1111/imr.13135 (2022).
Ni Choileain, S. et al. The dynamic processing of CD46 intracellular domains provides a molecular rheostat for T cell activation. PLoS ONE 6, e16287 (2011).
Ni Choileain, S. & Astier, A. L. CD46 processing: a means of expression. Immunobiology 217, 169–175 (2012).
Chang, C. H. et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013).
Cardone, J. et al. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat. Immunol. 11, 862–871 (2010).
Ni Choileain, S. et al. TCR-stimulated changes in cell surface CD46 expression generate type 1 regulatory T cells. Sci. Signal. https://doi.org/10.1126/scisignal.aah6163 (2017).
Arbore, G., Kemper, C. & Kolev, M. Intracellular complement — the complosome — in immune cell regulation. Mol. Immunol. 89, 2–9 (2017).
Arbore, G. & Kemper, C. A novel “complement-metabolism-inflammasome axis” as a key regulator of immune cell effector function. Eur. J. Immunol. 46, 1563–1573 (2016).
Ling, G. S. et al. C1q restrains autoimmunity and viral infection by regulating CD8+ T cell metabolism. Science 360, 558–563 (2018).
Paiano, J. et al. Follicular B2 cell activation and class switch recombination depend on autocrine C3ar1/C5ar1 signaling in B2 cells. J. Immunol. 203, 379–388 (2019).
Jimenez-Reinoso, A. et al. Human plasma C3 is essential for the development of memory B, but not T, lymphocytes. J. Allergy Clin. Immunol. 141, 1151–1154 e1114 (2018).
Kolev, M. et al. Diapedesis-induced integrin signaling via LFA-1 facilitates tissue immunity by inducing intrinsic complement C3 expression in immune cells. Immunity 52, 513–527.e8 (2020).
Liew, P. X. & Kubes, P. The neutrophil’s role during health and disease. Physiol. Rev. 99, 1223–1248 (2019).
Bashant, K. R. et al. Proteomic, biomechanical and functional analyses define neutrophil heterogeneity in systemic lupus erythematosus. Ann. Rheum. Dis. 80, 209–218 (2021).
Benoit, M. E., Clarke, E. V., Morgado, P., Fraser, D. A. & Tenner, A. J. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J. Immunol. 188, 5682–5693 (2012).
Baudino, L. et al. C3 opsonization regulates endocytic handling of apoptotic cells resulting in enhanced T-cell responses to cargo-derived antigens. Proc. Natl Acad. Sci. USA 111, 1503–1508 (2014).
Martin, M. et al. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ. 23, 903–911 (2016).
Tam, J. C., Bidgood, S. R., McEwan, W. A. & James, L. C. Intracellular sensing of complement C3 activates cell autonomous immunity. Science 345, 1256070 (2014).
Wang, Y., Tong, X., Zhang, J. & Ye, X. The complement C1qA enhances retinoic acid-inducible gene-I-mediated immune signalling. Immunology 136, 78–85 (2012).
Liszewski, M. K. & Atkinson, J. P. Membrane cofactor protein (MCP; CD46): deficiency states and pathogen connections. Curr. Opin. Immunol. 72, 126–134 (2021).
Schafer, N. et al. Complement factor H-related 3 enhanced inflammation and complement activation in human RPE cells. Front. Immunol. 12, 769242 (2021).
Kulkarni, H. S. et al. Intracellular C3 protects human airway epithelial cells from stress-associated cell death. Am. J. Respir. Cell Mol. Biol. 60, 144–157 (2019).
Chaudhary, N., Jayaraman, A., Reinhardt, C., Campbell, J. D. & Bosmann, M. A single-cell lung atlas of complement genes identifies the mesothelium and epithelium as prominent sources of extrahepatic complement proteins. Mucosal Immunol. 15, 927–939 (2022).
Sunderhauf, A. et al. GC1qR cleavage by caspase-1 drives aerobic glycolysis in tumor cells. Front. Oncol. 10, 575854 (2020).
Mahajan, S. et al. Local complement factor H protects kidney endothelial cell structure and function. Kidney Int. 100, 824–836 (2021).
Correa-Gallegos, D., Jiang, D. & Rinkevich, Y. Fibroblasts as confederates of the immune system. Immunol. Rev. 302, 147–162 (2021).
Friscic, J. et al. The complement system drives local inflammatory tissue priming by metabolic reprogramming of synovial fibroblasts. Immunity 54, 1002–1021 e1010 (2021).
Terri, M. et al. Mechanisms of peritoneal fibrosis: focus on immune cells-peritoneal stroma interactions. Front. Immunol. 12, 607204 (2021).
Shidham, V. B. The panorama of different faces of mesothelial cells. Cytojournal 18, 31 (2021).
Liu, Y. et al. Transition of mesothelial cell to fibroblast in peritoneal dialysis: EMT, stem cell or bystander? Perit. Dial. Int. 35, 14–25 (2015).
Lubbers, R., van Essen, M. F., van Kooten, C. & Trouw, L. A. Production of complement components by cells of the immune system. Clin. Exp. Immunol. 188, 183–194 (2017).
Markiewski, M. M. et al. C3a and C3b activation products of the third component of complement (C3) are critical for normal liver recovery after toxic injury. J. Immunol. 173, 747–754 (2004).
Yilmaz, M. et al. Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat. Neurosci. 24, 214–224 (2021).
Schafer, D. P. et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012).
Stevens, B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007).
Wang, C. et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 367, 688–694 (2020).
Kunz, N. & Kemper, C. Complement has brains — do intracellular complement and immunometabolism cooperate in tissue homeostasis and behavior? Front. Immunol. 12, 629986 (2021).
Tenner, A. J. Complement-mediated events in Alzheimer’s disease: mechanisms and potential therapeutic targets. J. Immunol. 204, 306–315 (2020).
Hong, S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016).
Brennan, F. H., Lee, J. D., Ruitenberg, M. J. & Woodruff, T. M. Therapeutic targeting of complement to modify disease course and improve outcomes in neurological conditions. Semin. Immunol. 28, 292–308 (2016).
Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016).
Jacob, A. & Alexander, J. J. Complement and blood–brain barrier integrity. Mol. Immunol. 61, 149–152 (2014).
Veerhuis, R., Nielsen, H. M. & Tenner, A. J. Complement in the brain. Mol. Immunol. 48, 1592–1603 (2011).
Datta, D. et al. Classical complement cascade initiating C1q protein within neurons in the aged rhesus macaque dorsolateral prefrontal cortex. J. Neuroinflammation 17, 8 (2020).
Ten, V. S. et al. Complement component c1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. J. Neurosci. 30, 2077–2087 (2010).
Meri, S. et al. Human protectin (CD59), an 18,000-20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology 71, 1–9 (1990).
Golec, E. et al. Alternative splicing encodes functional intracellular CD59 isoforms that mediate insulin secretion and are down-regulated in diabetic islets. Proc. Natl Acad. Sci. USA 119, e2120083119 (2022).
Golec, E. et al. A cryptic non-GPI-anchored cytosolic isoform of CD59 controls insulin exocytosis in pancreatic β-cells by interaction with SNARE proteins. FASEB J. 33, 12425–12434 (2019).
Krus, U. et al. The complement inhibitor CD59 regulates insulin secretion by modulating exocytotic events. Cell Metab. 19, 883–890 (2014).
King, B. C. et al. Complement component C3 is highly expressed in human pancreatic islets and prevents β cell death via ATG16L1 interaction and autophagy regulation. Cell Metab. 29, 202–210 e206 (2019).
Zelek, W. M., Xie, L., Morgan, B. P. & Harris, C. L. Compendium of current complement therapeutics. Mol. Immunol. 114, 341–352 (2019).
Schartz, N. D. & Tenner, A. J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflammation 17, 354 (2020).
Dijkstra, D. J., Joeloemsingh, J. V., Bajema, I. M. & Trouw, L. A. Complement activation and regulation in rheumatic disease. Semin. Immunol. 45, 101339 (2019).
Ghannam, A., Fauquert, J. L., Thomas, C., Kemper, C. & Drouet, C. Human complement C3 deficiency: Th1 induction requires T cell-derived complement C3a and CD46 activation. Mol. Immunol. 58, 98–107 (2014).
Ghannam, A. et al. Human C3 deficiency associated with impairments in dendritic cell differentiation, memory B cells, and regulatory T cells. J. Immunol. 181, 5158–5166 (2008).
Yan, B. et al. SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation. Sci. Immunol. https://doi.org/10.1126/sciimmunol.abg0833 (2021).
Afzali, B., Noris, M., Lambrecht, B. N. & Kemper, C. The state of complement in COVID-19. Nat. Rev. Immunol. 22, 77–84 (2022).
Bosmann, M. Complement control for COVID-19. Sci. Immunol. https://doi.org/10.1126/sciimmunol.abj1014 (2021).
Ma, L. et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci. Immunol. https://doi.org/10.1126/sciimmunol.abh2259 (2021).
Banda, N. K. et al. Analysis of complement gene expression, clinical associations, and biodistribution of complement proteins in the synovium of early rheumatoid arthritis patients reveals unique pathophysiologic features. J. Immunol. 208, 2482–2496 (2022).
Ellinghaus, U. et al. Dysregulated CD46 shedding interferes with Th1-contraction in systemic lupus erythematosus. Eur. J. Immunol. 47, 1200–1210 (2017).
Aragam, K. G. et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet. 54, 1803–1815 (2022).
Sunderhauf, A. et al. Regulation of epithelial cell expressed C3 in the intestine — relevance for the pathophysiology of inflammatory bowel disease? Mol. Immunol. 90, 227–238 (2017).
Satyam, A. et al. Intracellular activation of complement 3 is responsible for intestinal tissue damage during mesenteric ischemia. J. Immunol. 198, 788–797 (2017).
Roumenina, L. T., Daugan, M. V., Petitprez, F., Sautes-Fridman, C. & Fridman, W. H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 19, 698–715 (2019).
Reis, E. S., Mastellos, D. C., Ricklin, D., Mantovani, A. & Lambris, J. D. Complement in cancer: untangling an intricate relationship. Nat. Rev. Immunol. 18, 5–18 (2018).
Daugan, M. V. et al. Intracellular factor H drives tumor progression independently of the complement cascade. Cancer Immunol. Res. 9, 909–925 (2021).
Daugan, M. V. et al. Complement C1s and C4d as prognostic biomarkers in renal cancer: emergence of noncanonical functions of C1s. Cancer Immunol. Res. 9, 891–908 (2021).
Thurman, J. M. Complement and the kidney: an overview. Adv. Chronic Kidney Dis. 27, 86–94 (2020).
Richards, A. et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc. Natl Acad. Sci. USA 100, 12966–12971 (2003).
Sheerin, N. S. et al. Synthesis of complement protein C3 in the kidney is an important mediator of local tissue injury. FASEB J. 22, 1065–1072 (2008).
Brown, K. M. et al. Influence of donor C3 allotype on late renal-transplantation outcome. N. Engl. J. Med. 354, 2014–2023 (2006).
Portilla, D. & Xavier, S. Role of intracellular complement activation in kidney fibrosis. Br. J. Pharmacol. 178, 2880–2891 (2021).
Tziastoudi, M. et al. Key genetic components of fibrosis in diabetic nephropathy: an updated systematic review and meta-analysis. Int. J. Mol. Sci. https://doi.org/10.3390/ijms232315331 (2022).
Heurich, M. et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc. Natl Acad. Sci. USA 108, 8761–8766 (2011).
Samstad, E. O. et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J. Immunol. 192, 2837–2845 (2014).
Zimmer, S. et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci. Transl. Med. 8, 333ra350 (2016).
Bakke, S. S. et al. Cyclodextrin reduces cholesterol crystal-induced inflammation by modulating complement activation. J. Immunol. 199, 2910–2920 (2017).
Gerard, A., Cope, A. P., Kemper, C., Alon, R. & Kochl, R. LFA-1 in T cell priming, differentiation, and effector functions. Trends Immunol. 42, 706–722 (2021).
Seya, T. et al. CD46 (membrane cofactor protein of complement, measles virus receptor): structural and functional divergence among species (review). Int. J. Mol. Med. 1, 809–816 (1998).
West, E. E., Kunz, N. & Kemper, C. Complement and human T cell metabolism: location, location, location. Immunol. Rev. 295, 68–81 (2020).
Kremlitzka, M. et al. Alternative translation and retrotranslocation of cytosolic C3 that detects cytoinvasive bacteria. Cell Mol. Life Sci. 79, 291 (2022).
Russkamp, N. F. et al. Experimental design of complement component 5a-induced acute lung injury (C5a-ALI): a role of CC-chemokine receptor type 5 during immune activation by anaphylatoxin. FASEB J. 29, 3762–3772 (2015).
Tenner, A. J., Stevens, B. & Woodruff, T. M. New tricks for an ancient system: physiological and pathological roles of complement in the CNS. Mol. Immunol. 102, 3–13 (2018).
Ratajczak, M. Z. & Kucia, M. Hematopoiesis and innate immunity: an inseparable couple for good and bad times, bound together by an hormetic relationship. Leukemia 36, 23–32 (2022).
Elvington, M., Liszewski, M. K., Bertram, P., Kulkarni, H. S. & Atkinson, J. P. A C3(H20) recycling pathway is a component of the intracellular complement system. J. Clin. Invest. 127, 970–981 (2017).
West, E. E. & Kemper, C. Complement and T cell metabolism: food for thought. Immunometabolism 1, e190006 (2019).
Song, W. C. Crosstalk between complement and toll-like receptors. Toxicol. Pathol. 40, 174–182 (2012).
Barratt-Due, A., Pischke, S. E., Nilsson, P. H., Espevik, T. & Mollnes, T. E. Dual inhibition of complement and Toll-like receptors as a novel approach to treat inflammatory diseases — C3 or C5 emerge together with CD14 as promising targets. J. Leukoc. Biol. 101, 193–204 (2017).
Asgari, E. et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 122, 3473–3481 (2013).
Niyonzima, N. et al. Cholesterol crystals use complement to increase NLRP3 signaling pathways in coronary and carotid atherosclerosis. EBioMedicine 60, 102985 (2020).
Triantafilou, K., Hughes, T. R., Triantafilou, M. & Morgan, B. P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 126, 2903–2913 (2013).
Kumar, V. The complement system, toll-like receptors and inflammasomes in host defense: three musketeers’ one target. Int. Rev. Immunol. 38, 131–156 (2019).
Liu, H. et al. Mannan binding lectin attenuates double-stranded RNA-mediated TLR3 activation and innate immunity. FEBS Lett. 588, 866–872 (2014).
Acknowledgements
Work in the Kemper laboratory is supported in part by the Intramural Research Program of the National Institutes of Health, National Heart, Lung, and Blood Institute (ZIA/hl006223 to C.K.).
Author information
Authors and Affiliations
Contributions
All authors researched data for the article, made substantial contributions to discussions of the content and wrote, reviewed or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Nephrology thanks M. Bosmann, P. Cravedi, A. Jayaraman, L. Roumenina and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
West, E.E., Kemper, C. Complosome — the intracellular complement system. Nat Rev Nephrol 19, 426–439 (2023). https://doi.org/10.1038/s41581-023-00704-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41581-023-00704-1
This article is cited by
-
Canonical and non-canonical roles of complement in atherosclerosis
Nature Reviews Cardiology (2024)
-
The role of complement in kidney disease
Nature Reviews Nephrology (2023)
-
Senescent fibro-adipogenic progenitors are potential drivers of pathology in inclusion body myositis
Acta Neuropathologica (2023)
