
Abstract
Methylation quantitative trait loci (mQTLs) are essential for understanding the role of DNA methylation changes in genetic predisposition, yet they have not been fully characterized in East Asians (EAs). Here we identified mQTLs in whole blood from 3,523 Chinese individuals and replicated them in additional 1,858 Chinese individuals from two cohorts. Over 9% of mQTLs displayed specificity to EAs, facilitating the fine-mapping of EA-specific genetic associations, as shown for variants associated with height. Trans-mQTL hotspots revealed biological pathways contributing to EA-specific genetic associations, including an ERG-mediated 233 trans-mCpG network, implicated in hematopoietic cell differentiation, which likely reflects binding efficiency modulation of the ERG protein complex. More than 90% of mQTLs were shared between different blood cell lineages, with a smaller fraction of lineage-specific mQTLs displaying preferential hypomethylation in the respective lineages. Our study provides new insights into the mQTL landscape across genetic ancestries and their downstream effects on cellular processes and diseases/traits.
Similar content being viewed by others

Main
Methylation quantitative trait loci (mQTLs) are genetic variants that affect DNA methylation (DNAm) levels at CpG sites. Identifying and characterizing mQTLs is crucial to elucidating (1) the function of GWAS loci, (2) the role of DNAm as a potential causal mediator of genetic susceptibility and (3) the synergistic effect of genetic and environmental factors on disease risk.
MQTLs are abundant throughout the genome and they account for a substantial proportion of DNAm variation1,2,3,4,5,6,7,8,9,10,11,12,13,14,15. Current mechanistic hypotheses suggest that alterations in DNAm patterns can influence the three-dimensional structure of chromatin, thereby affecting the accessibility of regulatory regions to transcriptional machinery and other regulatory elements (REs)11. Additionally, DNAm changes may hinder or facilitate transcription factor (TF) binding, consequently influencing gene expression patterns. Furthermore, modifications to histone levels, induced by DNAm alterations, can lead to changes in chromatin structure and gene expression. It has also been proposed that the formation of trans-mQTL hotspots may be mediated by cis-eGenes2, which are genes located in close proximity to the mQTL region. However, our understanding of mQTLs is far from complete. First, well-sized mQTL studies have been conducted mainly in Europeans, with a few exceptions (for example, South Asians15). Second, methylomes have been primarily derived from whole blood DNA, or in only small numbers of purified cell subtypes; therefore, to which extent mQTLs are blood cell-subtype-specific remains unclear7,15. Third, while several mechanistic hypotheses provide potential insights into the relationship between DNAm and molecular processes, the precise mechanisms underlying mQTL formation and their impact on human complex traits at the population level are still not fully understood. In particular, a complete chain of evidence linking the molecular players in the path from DNA variation to human complex traits at the population level is still lacking.
Here we report a comprehensive identification and analysis of mQTLs in Chinese cohorts to address the above-mentioned questions.
Results
mQTL mapping, annotation and replication
We define mQTLs as single-nucleotide polymorphisms (SNPs) that can affect CpG DNAm levels, mCpGs as CpGs affected by mQTLs and associations as pairs of mQTLs and mCpGs. During the discovery phase, we examined 6.14 trillion associations between 7.58 million SNPs and 811,876 CpGs in the whole blood of 3,523 Han Chinese from the National Survey of Physical Traits (NSPT) cohort (Fig. 1a). This revealed 62.9 million study-wide significant associations (Fig. 1b), including 89.5% cis (P < 1.06 × 10−11), 3.6% long-rance cis (lcis) (P < 2.86 × 10−12) and 6.9% trans (P < 8.16 × 10−15) according to the previously proposed definitions (cis <1 Mbp, lcis 1–5 Mbp and trans >5 Mbp)2,5,10. Two-thirds (5.56 million) of all tested SNPs were identified as mQTLs (5.48 million cis, 271,994 lcis and 1.14 million trans), while one-third (284,128) of all tested CpGs were identified as mCpGs (267,891 cis, 7,746 lcis and 26,415 trans). After linkage disequilibrium (LD) pruning (r2 > 0.2), we obtained 859,089 cis-, 23,906 lcis- and 60,490 trans-mQTLs, which were more than double the numbers (394 K cis and 21 K trans) found using the 450 K array in 4,170 individuals of European ancestry from the Framingham Heart Study (FHS)2.

a, Diagram illustrating the overall study design. b, Plot showing all study-wide significant mQTL associations in NSPT: cis (<1 Mbp, P < 1.06 × 10−11, n = 56,289,777, red dots); lcis (1–5 Mbp, P < 2.86 × 10−12, n = 2,270,491, gray dots); trans (>5 Mbp or on different chromosomes, P < 8.16 × 10−15, n = 4,357,250, blue dots). All cis- and lcis- associations fall around the diagonal and so are hard to make out. SNP positions and background SNP number/Mbp (total = 7,576,990) are annotated on the x axis. CpG positions and background CpG number/Mbp (total = 811,876) are annotated on the y axis to make the variation in most of the background number/Mbp clearer. We restricted the y axes to lower values (10,000 for SNP and 1,900 for CpG) to avoid the domination by the human leukocyte antigen locus. c, Enrichment of mQTLs and their associated CpGs in functional elements (CTCF, E, P, PF and TFBS). Heatmap shows the fold changes of mQTLs and their associated CpGs compared with randomly selected (Methods) SNP–CpG pairs in all combinations of functional categories. One-tailed hypergeometric test is applied. The fold changes are labeled within each box. d, Enrichment of mQTLs and mCpGs in trait-associated sites in GWAS Catalog and EWAS Atlas compared with the randomly sampled sets (n = 104) from background. The x axis shows the proportion of trait-associated sites covered by mQTLs/mCpGs (blue lines), and randomly sampled sets (red histograms). e, Enrichment of mQTLs/mCpGs in different genomic regions compared with background. P values are from two-tailed hypergeometric tests and corrected by the Bonferroni method. Bonferroni-corrected *P < 0.05, **P < 10−10 and ***P < 10−50. f,g, Comparison of scaled effect sizes from study-wide significant mQTLs in NSPT and significant at FDR < 0.05 in CAS (f) or CGZ (g). ‘Cor’ is two-tailed Spearman’s rank correlation (both P < 1 × 10−323). ‘Dir’ means the proportion of allele effects in a consistent direction. CTCF, CTCF-enriched elements; E, enhancers; P, promoters; PF, promoter flanking regions; TFBS, TF binding sites.
The mQTL–mCpG pairs tend to be more enriched for TF binding sites, promoters and CTCF binding sites (Fig. 1c and Extended Data Fig. 1a–c) than distance-matched random pairs. The mQTLs showed significant enrichment of hematological traits in GWAS Catalog16 and mCpGs showed enrichment of various traits in EWAS Atlas17 (Fig. 1d and Supplementary Figs. 1–4).
Trans-mQTLs may have more direct biological significance than cis-mQTLs. This was supported by (1) the majority of trans-mQTLs (52.8% of 60,490 clumped trans-mQTLs) also being cis-mQTLs but not vice versa (3.7% of 859,089 clumped cis-mQTLs; Supplementary Fig. 5a); (2) a substantially higher proportion of trans-mCpGs also being cis-mCpGs (41.2% of 26,415) than vice versa (4.1% of 267,891; Supplementary Fig. 5b); (3) the explainable variance of mCpG DNAm decreased with LD (Supplementary Fig. 6); (4) trans-mQTLs showed a profound enrichment in promoters and exons compared to cis-mQTLs (Fig. 1e) and (5) trans-mCpGs showed a profound enrichment in TSS200 and CpG islands compared to cis-mCpGs (Fig. 1e). Interestingly, genes in the vicinity of cis-mQTLs or cis-mCpGs were more enriched for basic functional processes, whereas genes in the vicinity of trans-mQTLs or trans-mCpGs were more enriched for immune-related ontologies and pathways (Supplementary Figs. 7 and 8). Otherwise, no noticeable differences in methylation levels, methylation variance and heritability were observed among cis-, lcis- and trans-mCpGs (Supplementary Fig. 9).
We replicated the 62.9 million study-wide significant associations in two independent Han Chinese cohorts (Chinese Academy of Sciences (CAS) cohort, n = 1,060 and clinical trials of chiglitazar (CGZ), n = 798). Replication rates were high in both CAS (93.8%) and CGZ (87.1%, false discovery rate (FDR) < 0.05), with high levels of directional consistency (99.7% in CAS and 99.8% in CGZ) and highly consistent allele effect sizes (Spearman correlation, r = 0.97 in CAS and r = 0.98 in CGZ; Fig. 1f,g).
East Asian mQTLs
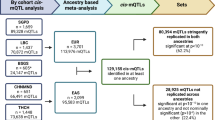
We compared our mQTLs with those reported in the recent meta-analysis of European cohorts from the Genetics of DNA Methylation Consortium (GoDMC)8 and found that 64.7% were shared between the two datasets. The larger number of mQTLs found in GoDMC (3.46 million in GoDMC versus 2.65 million in NSPT; Methods) is likely due to its larger sample size. The mQTLs identified in the two studies showed similar minor allele frequency (MAF) distributions in their respective populations (Fig. 2a and Supplementary Tables 1 and 2). However, the likelihood of an mQTL in one study being significant in another study heavily depended on its MAF in the other study, with low MAF values being substantially less likely (Fig. 2a). We identified 248,173 East Asian (EA)-specific mQTLs that were skewed toward lower MAFs in Europeans and had higher MAFs in EAs (Supplementary Fig. 10a). Most of the EA-specific mQTLs were replicated in the CAS cohort, but only a small percentage (12.8%) was observed in FHS (Supplementary Table 3).

a, The left and right panels show the replication rates (redness) and numbers (square size) of study-wide significant mQTLs in GoDMC and NSPT within different SNP MAF bins, respectively. The dependence on MAF in the latter is particularly evident in the low MAF bins (middle panels). b, Scatter plot showing 541 EA-specific trans-colocalizations (36 loci and 15 traits). The x axis indicates the SNP position; the y axis indicates the position of colocalized mCpGs in the locus. Points represent the 541 trans-colocalizations. The color of points indicates different traits, with the size indicating the significance of SMR test (−log10 of SMR P values). c, A schematic diagram of hematopoietic differentiation showing the regulation of different genes during hematopoietic cell differentiation by the heptad complex comprising ERG, TAL1, RUNX1, LYL1, LMO2, GATA2 and FLI1. d, A protein–protein interaction network (STRING V11.5; Cytoscape V3.9) of ERG and the 62 TFs for whose motifs the 233 CpGs trans-colocalized at chr21q22.2 loci were substantially enriched. e, Enrichment of the chr21q22.2 trans-colocalization-related genes (ERG, 62 TFs and 195 annotated genes of trans-colocalized CpGs) in biological pathways and processes (Metascape v3.5). X axis indicates −log10(Q value) that was obtained from a one-tailed hypergeometric test.
Of the 2.65 million mQTLs in NSPT, 39,162 overlapped with signals in the GWAS Catalog (P < 5 × 10−8), accounting for 47.5% of the total 82,392 GWAS signals examined (Supplementary Table 4). The EA-specific mQTLs accounted for 3.0% and 0.94% signals in Biobank Japan (BBJ) and UK Biobank (UKBB), respectively (Supplementary Fig. 10b and Supplementary Table 4). EA-specific mQTLs showed a stronger overlap with EA-specific GWAS signals, explaining 4.8% BBJ-specific GWAS signals compared to only 0.81% UKBB-specific GWAS signals, with representing a 6.16-fold improvement in efficiency (95% confidence interval (CI): 5.90–6.43; Supplementary Fig. 10c and Supplementary Table 5). This is again largely explained by allele frequency differences (Supplementary Fig. 10d).
A colocalization analysis of 248K EA-specific mQTLs and all 230 GWASs in BBJ identified 152 mQTLs in 44 distinct loci (>1 Mbp) showing significant (PSMR < 3.7 × 10−9 and PHEIDI > 0.05) evidence of colocalizations involving 33 traits (Supplementary Table 6). Only three of these loci could be further supported by eQTLs in peripheral blood of 298 Japanese individuals in Human Genetic Variation Database (HGVD)18,19 (PeQTL < 1 × 10−5). Eight loci showed significant colocalizations with 20 CpGs in NSPT and adult height in BBJ (PSMR < 1.25 × 10−10, PHEIDI > 0.05). One locus located in the intron of ELF1 (13q14.11; Extended Data Fig. 2a) showed significant associations with seven cis-mCpGs in NSPT (PmQTL < 8.81 × 10−110), adult height in BBJ (PGWAS = 2.1 × 10−11, PSMR < 1.25 × 10−10 and PHEIDI > 0.05), and the expression of ELF1 in the peripheral blood of the Japanese sample (eQTL PSMR = 5.88 × 10−13), suggesting a role of CpGs in mediating genetic association of adult height. ELF1 variants have been shown to have a large effect on adult height in EA populations20,21 with rs7335629 being an EA-specific signal in the latest human stature study21. This SNP and one of its associated CpG (cg21067652) are predicted to be in co-opening regions with binding sites from the same TFs (Extended Data Fig. 2b,c). A two-sample MR analysis revealed a causal effect of cg21067652 on ELF1 expression and adult height (Extended Data Fig. 2d,e). Therefore, the colocalization of EA-specific mQTLs and trait GWASs enhances our understanding of trait associations at the epigenetic level.
EA-specific cis-colocalizations
Cis- and trans-mQTLs were separately analyzed for colocalization with 107 overlapping GWASs between BBJ and UKBB. The cis-analysis identified 216 distinct loci (>1 Mbp) showing significant colocalizations (PSMR < 3.5 × 10−9 and PHEIDI > 0.05) between cis-mQTLs in NSPT and 45 GWASs in BBJ. Among these, 96 loci were colocalized with 38 GWASs exclusively in EAs (Supplementary Table 7 and Supplementary Fig. 11a). The most frequently associated phenotypes were cardiometabolic phenotypes (13/38), followed by hematological phenotypes (8/38). The most significant colocalization was identified for an intergenic variant at 12q24.13 (rs4534647, PSMR = 7.0 × 10−32; Supplementary Fig. 11b), which was cis-associated with a CpG in the first intron of MAPKAPK5 (cg22778180, PmQTL = 2.2 × 10−110) in NSPT and was associated with mean corpuscular volume (PGWAS = 1.9 × 10−43), red blood cell count (PGWAS = 4.4 × 10−25) and alanine aminotransferase (PGWAS = 4.8 × 10−23) in BBJ. The T allele of rs4534647 is common in EAs (f = 0.62) but low frequent in Europeans (f = 0.01; Supplementary Fig. 11c), explaining the lack of mQTL, genetic association and colocalization in Europeans.
EA-specific trans-colocalizations
The trans-analysis identified 46 loci showing significant trans-colocalizations with 23 distinct GWASs in BBJ. Of these loci, 36 exhibited EA-specific trans-colocalizations involving 486 independent mCpGs and 15 GWASs of primarily hematological traits (8 of 15; Supplementary Table 8 and Fig. 2b). Compared to nonspecific colocalizations, EA-specific ones were significantly enriched for trans (Fisher’s exact test, odds ratio (OR) = 4.48, 95% CI (2.05,10.65); Extended Data Fig. 3a,b) and EA-specific trans-colocalization were significantly enriched in predicted transcriptional and enhancer regions (P < 5 × 10−8; Extended Data Fig. 3c,d), emphasizing their population-specific and functional significance.
The most significant EA-specific trans-colocalization was located in the intron of ERG on chr21q22.2 (rs80109907, in complete linkage with rs77106233 in hotspot H16, PSMR = 7.9 × 10−35; Table 1 and Supplementary Fig. 11d), where the A allele was primarily positively (97%) trans-associated with 233 independent mCpGs and positively associated with basophil count in BBJ (PGWAS = 1.1 × 10−59). The A allele is common in EA populations (f = 0.11) but rare in European populations (f = 0.01; Extended Data Fig. 4a). rs80109907 was significantly associated with eosinophils, monocytes, red blood cells, neutrophils and platelets (Supplementary Table 9 and Extended Data Fig. 4b). A summary-data-based mendelian randomization (SMR) analysis revealed weaker (compared to basophil count) but significant trans-colocalizations involving several blood cell count and immune-related diseases, including monocytes, eosinophils, white blood cells, urticaria, pericarditis and asthma (Supplementary Table 10 and Extended Data Fig. 4c). A two-sample MR analysis further identified 39 causal CpGs for these traits (Extended Data Fig. 4d). The trans-mCpGs were significantly enriched in motifs of 62 TFs (Supplementary Table 11), with 13 also appearing in blood cell TF ChIP–seq data (Supplementary Table 12). TAL1 and RUNX1, two of these TFs, interact directly with ERG and, together with ERG and four other proteins, form a TF heptad complex that has an important role in the transcriptional regulation of hematopoietic stem cells22,23 (Fig. 2c). Protein interaction analysis revealed that these 62 TFs and ERG formed a large protein interaction network (Fig. 2d). GO analysis showed that these TFs, together with the genes in the vicinity of the mCpGs, were significantly enriched in hematopoiesis and regulation of leukocyte differentiation (Fig. 2e). These results support that trans-regulated DNAm changes affect the binding efficiency of multiple TFs in the ERG protein complex, further regulating the process of hematopoietic cell differentiation.
Most mQTLs are not cell-type or cell lineage specific
An important yet unresolved question relates to whether mQTLs are present in all underlying blood cell types or only in specific subsets. While addressing this in a mixed cell-type tissue like blood is subject to limitations, we nevertheless applied CellDMC24, an algorithm designed to detect cell-type-specific differential DNAm, to identify cell-type-specific mQTLs among the mQTLs detected in NSPT (Methods). Running CellDMC at the resolution of lymphoid and myeloid lineages, and separately again at the higher resolution of six cell types, we identified many lineage- and cell-type-specific mQTLs (Supplementary Fig. 12a,b). We verified using Monte-Carlo analysis that the number of lineage and cell-type-specific mQTLs identified on random SNP–CpG pairs using CellDMC is negligible (Supplementary Table 13). We observed that many mQTLs significant in one lineage also displayed associations in the other, suggesting that most mQTLs are lineage-independent. To independently confirm this, we extended previous simulation models25 to estimate the sensitivity of detecting lineage-independent mQTLs simultaneously in the myeloid and lymphoid lineages (Methods). We estimated approximately 93% shared mQTLs between lineages (Fig. 3a,b), consistent with previous lower bound (that is, 70%) from BLUEPRINT7,26. Cell-lineage/cell-type-specific mQTLs were validated in the independent Han Chinese cohort (CGZ; Fig. 3c,d), as well as in sorted immune-cell subsets from BLUEPRINT (Fig. 3e). Scatterplots of DNAm versus myeloid or lymphoid fraction, stratified by genotype, provided visual confirmation of myeloid-specific, lymphoid-specific and lineage-independent mQTLs (Fig. 3f). The effect size of myeloid-mQTLs was significantly larger than that of lymphoid mQTLs for both cis- and trans-mQTL categories (Supplementary Fig. 12c,d), consistent with myeloid cells being the dominant component in blood. mQTLs were then stratified into four groups (lineage-independent, myeloid-specific, lymphoid-specific and rest) and tested separately for enrichment of myeloid- and lymphoid-specific hypomethylated differentially methylated CpGs (DMCs; Methods). Myeloid-specific and lymphoid-specific mQTLs were strongly enriched for corresponding myeloid-hypomethylated DMCs and lymphoid-hypomethylated DMCs (Fig. 3g). Interestingly, using eFORGE2, which tests for enrichment of DNase hypersensitive sites (DHSs) in myeloid and lymphoid cell types from ENCODE, BLUEPRINT and the Epigenomic Roadmap, lineage specificity was only observed for myeloid-mCpGs (Methods; Fig. 3h and Supplementary Fig. 13), probably because DHS displays lower cell lineage specificity than differential DNAm.

a, Flowchart describing the procedure for estimating the fraction of lineage-independent mQTLs (fShared_mQTLs). The sensitivity to detect a shared mQTL in the myeloid and lymphoid lineages is estimated via a mathematical integration of the observed effect sizes and the corresponding sensitivities as derived from a simulation model (Methods). b, Plots of sensitivity (SE) to detect a shared mQTL in each of the myeloid and lymphoid lineages versus variance-adjusted effect size (top, cumulative SE), as inferred from a realistic simulation model. Bottom: variance-adjusted effect size distribution. c,d, Scatterplots display the t-statistics of association between genotype and DNAm in six cell types (c) and two cell lineages (d) in the discovery cohort (NSPT, n = 3,523, x axis) versus the corresponding statistics in the validation cohort (CGZ, n = 798, y axis). Fisher’s exact test OR and two-tailed P value are given. e, Scatterplots display the signed −log10(P) of myeloid or lymphoid mQTLs (NSPT, n = 3,523, x axis) versus the corresponding values in the purified samples from BLUEPRINT (n = 197, y axis). The best fit (red lines) and 95% CI (gray bands) are given, with P values from linear regressions. Barplots indicate the overlap of myeloid/lymphoid mQTLs with those identified in BLUEPRINT against the expected number under the null with error bar indicating 95% CI. One-tailed binomial P value is given. f, Scatterplots of DNAm (β; y axis) versus cell-type fraction F (x axis) for four mQTLs with samples colored by genotype. The top two mQTLs are examples of myeloid- and lymphoid-specific mQTL, with the x axis labeling the myeloid and lymphoid fraction, respectively. The bottom two mQTLs are examples of two cell-lineage-independent mQTLs, with the left mQTL being equally dominant in myeloid and lymphoid subsets and the right mQTL being more dominant in the myeloid subset. g, Enrichment analysis of all myeloid and lymphoid-specific mCpGs among myeloid and lymphoid-specific hypomethylated DMCs. ORs and P values derive from one-tailed Fisher’s exact tests. h, eFORGE analysis of top 1,000 myeloid and lymphoid mCpGs. Y axis indicates −log10(P) from one-tailed hypergeometric test of myeloid and lymphoid mCpGs for DHSs as derived in lymphoid and myeloid cell types (x axis) from BLUEPRINT, ENCODE, consolidated Roadmap (ERC) and original Roadmap (ERC2012). The color of the cell type indicates its source. Overall results are very similar when using the top-10,000 myeloid and lymphoid mCpGs. Th1: type 1 T helper cell; Th2: type 2 T helper cell; macrophage LPS: lipopolysaccharide-induced macrophage; Neut, neutrophil; Mono, monocytes; CD4T, CD4+ T cells; NK, natural killer; Mye, myeloid lineage; Lym, lymphoid lineage.
Regulatory network explains >40% cis-mQTLs
We then investigated the proportion of mQTLs explained by chromatin interaction regions and to what extent they contribute to gene regulation. The results showed that 72.3% of cis-mQTLs and their mCpGs were located within the same topologically associating domains (TADs), representing a 1.6-fold enrichment over distance-matched SNP–CpG pairs (Methods; Fig. 4a and Extended Data Fig. 1d). The cis-mQTLs and their mCpGs were also more prominent in PCHi-C loops, HiChIP loops (Fig. 4b and Extended Data Fig. 1e), co-opening region and co-active region (Fig. 4c). An OpenCausal database27 analysis showed significant enrichment of our cis/lcis-mQTLs in blood-specific SNPs linked to chromatin accessibility (Fig. 4d). Next, we considered two possible mechanistic models (M1 and M2; Methods; Fig. 4e,f). M1 assumes that a mQTL in a RE alters chromatin accessibility, for example, by affecting pioneer factor binding. This could further influence DNAm of distal CpGs through 3D interactions (Fig. 4e). In M2, a RE does not necessarily change chromatin accessibility but influences DNAm at a distal mCpG by disturbing the binding affinity of cofactors or nonpioneer TFs (Fig. 4f). The analysis showed that a total of 40.4% cis/lcis-mQTLs could be explained by our proposed models (38.1% M1 only, 7.9% M2 only, 40.4% M1 + M2; Fig. 4g).

a, Top: the schema of mQTL and its associated CpG located within/across/outside a TAD. Bottom: comparison of the pairs of mQTL and its associated CpG within/across/outside TADs with two control groups. b, Top: the schema of mQTL and its associated CpG validated by a chromatin loop. Bottom: comparison of the pairs of mQTL and its associated CpG validated by PCHi-C or HiChIP loops with two control groups. c, Top: the schema of mQTL and its associated CpG being co-opening or co-active. Bottom: comparison of the pairs of mQTL and its associated CpG being co-opening/co-active with two control groups. d, Enrichment of mQTLs in OpenCausal variants of blood tissue derived from an external database (two-tailed Fisher’s exact test, P = 1.6 × 10−16, 2.02-fold). e, Schematic overviews of the M1 regulatory process. f, Schematic overviews of the M2 regulatory process. g, Proportion of mQTLs interpreted by the two mechanisms M1 and M2.
mQTL hotspots are mediated by TFs
Our 1.14 million trans-mQTL SNPs were assigned to 1,727 distinct loci with a minimum distance of at least 1 Mbp between them (Methods). Of these, 959 (55.5%) were considered mQTL hotspots, which contained mQTLs trans-associated with multiple independent mCpGs, ranging from 776 (44.9%) with two independent mCpGs, 16 (0.9%) with >100 independent mCpGs (H1–H16), up to with 692 independent mCpGs (Fig. 5a and Supplementary Table 14). Of the 16 hotspots, 16 were replicated in CAS, and eight were found in the 22 hottest hotspots in FHS2. Except for H9, all 16 hotspots contained TF genes, and 10 had significant evidence for their trans-mCpGs being enriched in the motifs of corresponding TFs (Table 1). Defining an index mQTL as the mQTL having the largest number of independent trans-mCpGs in a hotspot, we found that its allelic effect on trans-mCpGs tended to be in the same direction (Table 1). In 12/16 hotspots, we found the index mQTLs or their high LD SNPs (r2 > 0.6; Supplementary Table 15) in GWAS Catalog loci. The associated traits were predominantly hematological (10/12) and inflammatory traits (8/12), further supporting the role of trans-mQTLs in maintaining blood cell identity.

a, The number of independent trans-mCpGs associated with each trans-mQTL identified in NSPT (y axis). The 16 mQTL hotspots (>100 independent trans-mCpGs) are labeled by ‘H1’–‘H16’. Single asterisk indicates the hotspots also reported in the FHS cohort. b, The number of independent trans-mCpGs associated with each trans-mQTL (red points, left y axis) and the number of TFs near each trans-mQTL (<1 Mbp, blue points, right y axis) for each trans-mQTL on chr19 (x axis indicates the position of each trans-mQTL on chr19). Correlation and P value from two-tailed Spearman’s rank correlation test between the two numbers are given. c, Relationship between the proportion of trans-mQTLs located near a TF (<1 Mbp) and the number of associated independent trans-mCpGs. d, OR and P value from two-tailed Fisher’s exact test showing the likelihood of being trans-mQTLs for the SNPs near a TF (<1 Mbp) versus that not near a TF. e, Plot indicates trans-mQTL hotspots enrichment in super enhancer, TAD, HiChIP promoter–promoter interaction (HiChIP Prom–Prom) and promoter–other interaction (HiChIP Prom–other). P values from one-tailed hypergeometric tests are given. f, Enrichment of annotated genes of trans-mCpGs associated with 16 hotspot index mQTLs in biological processes. The enrichment results of annotated genes of trans-mCpGs associated with the index mQTLs in biological processes are compared with all trans-mCpGs. There were in total 58 significant (FDR < 0.05) terms, simplified by clustering on semantic similarity. Left is the heatmap of the similarity matrix of the 58 terms, and the word cloud annotations that summarize the functions with keywords in every cluster are shown on the right side of the heatmap. The font size of the keywords corresponds to the enrichment significance of the keywords compared to the background (biological process) vocabulary. Only the clusters of word cloud with a size of at least 5 are shown.
Chr19 had the highest density of mQTL hotspots (0.58/Mbp) and TF gene density (5.6 TFs/Mbp) among all chromosomes, followed by chr17 with the second highest density of mQTL hotspots (0.43/Mbp) and the second highest TF gene density (0.9 TFs/Mbp; Supplementary Table 16). Notably, the hotspots on chr19 were predominantly located in the high-density TF regions (Fig. 5b). The hotspot density was significantly correlated with the TF density across all chromosomes, with (Spearman correlation, r = 0.71, P = 1.23 × 10−4) or without (r = 0.66, P = 5.52 × 10−4) chr19 (Supplementary Table 16). Trans-mQTLs were more likely to be located near a TF gene than a random SNP in the genome (OR = 1.53; Fig. 5c,d). These findings underscore the significant role of TFs in the formation of trans-mQTLs and mQTL hotspots throughout the genome.
Trans-mQTL hotspots marked with super enhancers
We found all 16 hotspots identified in our data contained super enhancers but not all had TF genes (Supplementary Tables 17–19). Super enhancers are characterized as a cluster of tightly connected enhancers located in a relatively larger chromatin region that bind by key TFs and mediators, to drive high expressions of their associated genes28,29. Trans-mQTL hotspots were also enriched with chromatin interactions (promoter–promoter and promoter–other interaction defined using PCHi-C and HiChIP) and accessibility-associated variants (Fig. 5e and Supplementary Table 20), which was consistent with the association of super enhancers with high histone modification levels, DNase I hypersensitivity and chromatin interactions28,29. Additionally, trans-mQTL hotspots and their associated CpGs were enriched within genes that are involved in cellular processes (for example, transport, signaling, cell communication, adhesion and morphogenesis) and cell components (for example, plasma membrane, cell periphery, cell junction and projection; Fig. 5f), which was in concordance with the role of super enhancers and their associated genes in maintaining cell identity28,29. These findings suggested that super enhancers have key roles in the formation of trans-mQTL hotspots.
A FOSL2 hotspot influences eosinophil count
We observed a significant enrichment of trans-mCpGs in the binding sites of the TFs located near their respective trans-mQTLs (Table 1). These TFs are often known susceptibility loci of human traits and diseases (Supplementary Fig. 14), suggesting a functional role of trans-mCpGs as a potential causal mediator of genetic susceptibility. To further investigate this, we used H2 (the most significant from TFmotifView) and H5 (the most significant from PWMEnrich) as examples (Table 1). The G allele of the index mQTL rs4666078 at H2 was nominally significantly (P = 4.3 × 10−3) associated with a reduced eosinophil count in a recent GWAS of eosinophil count in EA30 and led to a decreased DNAm at all its 232 trans-mCpGs from 204 distinct loci throughout the genome (Fig. 6a). The trans-mCpGs were significantly enriched for FOSL2 motifs (76.7%, P = 2.50 × 10−177; Table 1) and FOSL2 ChIP–seq binding sites (78.0%, P = 1.25 × 10−101; Fig. 6a). The 151 genes in the vicinity of the 232 trans-mCpGs were significantly enriched in eosinophil count in DisGeNET (Fig. 6b). Additionally, these trans-mCpGs were significantly enriched in genome-wide significant CpGs identified by the to-date only EWAS of tissue eosinophilia (P = 2.19 × 10−24; Fig. 6c), in which 46/232 (19.8%) were genome-wide significant, and for 39/46 (84.8%), the G allele-induced decrease in DNAm was associated with a reduced risk of tissue eosinophilia (Supplementary Fig. 15). SMR and heterogeneity in dependent instrument test (HEIDI) analysis showed nominally significant colocalization between most of the 232 trans-mCpGs and eosinophil count (100% PSMR < 0.05, 99% PHEIDI > 0.05). Two-sample MR analysis identified 21/232 CpGs as putative causal factors of blood eosinophil count (Fig. 6d and Supplementary Table 21), and for all 21 CpGs, G-induced decrease in DNAm led to a reduced eosinophil count. These included cg22652934 at RUNX1, which encodes a TF well-known to be functionally involved in the specification of myeloid and lymphoid cell lineages from hematopoietic stem cells31. These results suggest that the FOSL2-hotspot may modulate eosinophil count via DNAm at multiple trans-mCpGs.

a, Circos plot displaying the enrichment of the 232 trans-mCpGs associated with the index SNP rs4666078 (at the hotspot H2) for FOSL2 ChIP–seq binding sites. The inner (orange/gray) link lines show the 232 trans-mCpGs, and the orange ones represent those overlapped with FOSL2 binding sites. The inner red barplots show −log10(Q) of peak-caller MACS2 indicating ChIP–seq signals score (all Q < 1 × 10−5). The outer blue barplots show −log10(P) of trans-mQTL associations. Outer track represents the directionality of DNAm change (rs4666078-G allele) of the trans-mCpGs: cyan (hypermethylation) and magenta (hypomethylation). b, Enrichment of the 151 genes annotated to the 232 trans-mCpGs for human diseases and traits based on DisGeNET knowledgebase compared with all human genes (according to the NCBI). Only significant results are shown (one-tailed Fisher’s exact test, FDR < 0.05). c, Top: the enrichment (one-tailed hypergeometric test) of the 232 trans-mCpGs in 24,114 genome-wide significant CpGs associated with tissue eosinophilia (850 K array, Korean population, n = 147; Supplementary Protocols) compared to background. Bottom: the association significance with tissue eosinophilia (y axis) of the 46/232 mCpGs (red) and background (blue). P value from one-tailed Kolmogorov–Smirnov test is given. d, Two-sample MR results showing 21 CpGs are causal for eosinophil count in whole blood (FDR < 0.05). The left and right y axes indicate the CpGs and their genomic annotations. Each point and the error bar indicate the causal effect (β from two-tailed MR–IVW test) and its 95% CI. Tissue eosinophilia (+) means that CpG methylation is positively associated with the risk of tissue eosinophilia, DisGeNET means that CpG-annotated gene is related with eosinophil count in DisGeNET and other means that the CpG or its annotated gene is not reported in either dataset.
An NFKB1 hotspot may mediate the risk of obesity
H5 on chr4 represents our most significant finding from PWMEnrich (Table 1). The index mQTL rs3774937 at H5 was significantly associated with ulcerative colitis in GWAS Catalog (P = 5.0 × 10−8). Relative to the T allele, the C allele of rs3774937 led to a decreased DNAm at 99.1% of its 448 study-wide significant (P < 8.16 × 10−15) trans-mCpGs from 336 distinct loci throughout the genome (Table 1), which is largely consistent with the findings in 3,841 Dutch Europeans1. The rs3774937 in the first intron of NFKB1 is a cis-eQTL of NFKB1 (P = 7.6 × 10−53, eQTLGen whole blood). Under a more relaxed significance threshold (P < 1 × 10−8), rs3774937 had 856 trans-mCpGs, which were significantly enriched for NFKB1 motifs (63%) and NFKB1 ChIP–seq binding sites (36.6%; Fig. 7a,b). Given that a recent EWAS for body mass index (BMI) has revealed a strong enrichment for the NFKB-pathway32, we hypothesized that some of the NFKB1 trans-mQTLs may be associated with BMI. To test this in our Han Chinese cohort, we first established that of 364 CpGs known to be associated with BMI32,33,34,35, 267 did so also in our Asian cohort (Fig. 7c). We observed that 15 of the 364 BMI CpGs were NFKB1 trans-mQTLs (Fisher’s exact test, P = 2.7 × 10−19), and that NFKB1 trans-mQTLs displayed a very strong association with BMI in our Chinese cohort, as well as in an independent European cohort36 (Fig. 7d,e). Although SMR–HEIDI analysis of the NFKB1 hotspot was nonsignificant, due to the lack of association between the index SNP and BMI, this is complicated by the trait’s high polygenicity and the high degree of the index SNP’s pleiotropy. Hence, we asked if NFKB1 trans-mQTLs enriched for NFKB1 binding motifs/sites, which are also associated with BMI in our cohort, displayed any evidence of being causal mediators of BMI. Using a two-step MR strategy (Methods), we observed stronger evidence for DNAm levels at NFKB1 trans-mQTLs being causal mediators for BMI, as opposed to being a consequence of BMI (Fig. 7f and Supplementary Table 22). In contrast, when considering the BMI-associated CpGs (excluding the subset of NFKB1 trans-mQTLs), we observed stronger evidence for DNAm at these sites being a consequence of BMI (Fig. 7g and Supplementary Table 23), consistent with previous observations32. This suggests that DNAm at a subset of NFKB1 binding targets may be causal for high BMI, with a component of variation at these loci being under genetic influence. We next reasoned that if DNAm at these loci mediates the risk of obesity, these loci may also display a significant interaction between genotype and BMI that better models their DNAm variation. We were able to confirm a number of NFKB1 trans-mQTLs that displayed a significant interaction between genotype and BMI (Fig. 7h). Of note, among the three CpGs exhibiting both causal and interaction effects, one annotated to PTPN3, a protein-tyrosine-phosphatase gene that has been linked causally to obesity37, and another to NOD2, an intracellular innate immunity protein gene that has been shown to be protective of diet-induced obesity and colitis38,39,40, which also has polymorphisms associated with inflammatory bowel disease41. Other genes highlighted by our interaction analysis have also been previously implicated in obesity, inflammatory bowel disease or type 2 diabetes, including FOXP1 (ref. 42), CDKAL1 (refs. 43,44,45), HSD17B12 (ref. 46), ALAS1 (ref. 47), COL15A1 (ref. 48) and DNAH10 (ref. 49).

a,b, Circos plots displaying the enrichment of NFKB1 binding motifs (a) and NFKB1 ChIP–seq binding sites (b) among trans-mCpGs associated with the SNP linked in cis with NFKB1. The inner track labels the enriched trans-mCpGs (red). Blue barplots display the −log10(P) of trans-mCpGs. Red barplots display corresponding −log10(P) of ChIP–seq peak or TF motif enrichment from one-side Fisher’s exact test. The outer track represents the directionality of DNAm change of the trans-mCpGs: cyan (hypermethylation) and magenta (hypomethylation). c, Scatterplots displaying the signed −log10(P) of 364 BMI-CpG associations reported in the literature (y axis) against their corresponding values in NSPT (x axis). The best fit (red line) and 95% CI (gray bands) are given, with P values from linear regressions. d, Association of NFKB1 trans-mCpGs with BMI in two independent cohorts (NSPT and Buccal IC). Plots display the −log10(P) of CpG-BMI association (y axis) versus the CpG index position (x axis) for the NFKB1 trans-mCpGs (red line) versus that of randomly selected CpGs (blue). P value is from a one-tailed Binomial test. e, Scatterplots displaying the t-statistics of association of DNAm of NFKB1 trans-mCpGs with risk allele (y axis) versus their corresponding t-statistics of association of DNAm with BMI in the two independent cohorts (x axis). f, MR for the 35 NFKB1 trans-mCpGs associated with BMI. X axis labels −log10(FDR) from two-tailed MR–IVW test. Major color indicates direction of effect, with left panel displaying results for model where DNAm mediates the effect of SNP on obesity and right panel is for model where obesity affects DNAm. g, similar to f, but for BMI-associated CpGs reported by BMI EWAS studies. h, Scatter plot displaying the interaction between BMI and genotype on BMI-associated NFKB1 trans-mCpGs. X axis labels −log10(P) of the univariate association between DNAm and genotype, y axis labels −log10(P) of the univariate association between DNAm and BMI. The color of point indicates −log10(P) of the interaction term between genotype and BMI.
Discussion
This work advances our understanding of mQTLs in several ways. First, using the 850 K beadarray, we identified more than double the number of mQTLs than previous studies and integrated them into a pan-ancestry mQTL database that also includes published European and South Asian data (https://www.biosino.org/panmqtl/). While the majority of mQTLs are shared between EA and European populations, a significant proportion are specific to EAs, improving functional annotation of GWAS findings. Colocalization of cis-mQTLs with GWASs facilitates fine-mapping of trait-variants, while trans-colocalizations help pinpoint biological pathways contributing to variant-trait association, revealing the role of methylation in these pathways. For instance, we identified an EA-specific trans-colocalization mQTL network involved in basophil differentiation by affecting the binding efficiency of the ERG protein complex.
Second, our results indicate that blood cell-subtype-specific mQTLs are relatively uncommon, in line with recent findings15,50, but that the smaller number of lineage-specific mQTLs are more likely to map to hypomethylated cell-lineage-specific marker genes. Consistent with this, TFs implicated in mQTLs (for example, NFKB1 or CTCF) are generally not immune-cell-type specific, displaying variable but consistently nonzero expression among such cell subtypes. Simulation, however, indicates that much larger studies will be needed to ascertain in which cell types a given mQTL is truly not present in25.
Third, we proposed two possible mechanistic models that together explain over 40% cis-mQTL-CpG associations. Of these, the M1 model accounted for a relatively large proportion of the explanation, suggesting that the influence of an mQTL on DNAm is directly related to pioneer TF binding affinity. It also suggests that super enhancers have a key role in the formation of trans-mQTL hotspots. The enrichment of variants related to blood cell traits in trans-mQTL hotspots also supports the hypothesis that these hotspots have key roles in maintaining blood cell identity. In addition to TFs, other DNA binding factors such as TETs and DNMTs can also give rise to mQTLs. The models proposed here are rather simple, and more complex models can be developed to explore the mechanism of mQTLs.
Fourth, our trans-mQTL hotspot analysis has shown that many key hotspots (for example, NFKB1 and FOSL2) are independent of ethnicity. The hotspot associated with FOSL2 potentially modulates eosinophil count via the TF RUNX1, which also shows a degree of myeloid-specificity. Besides the potential role of the NFKB1 hotspot in mediating the risk of ulcerative colitis1, our analyses have revealed a role in obesity. MR on a subset of NFKB1-associated trans-mCpGs revealed that these could be causally implicated in mediating the risk of obesity, in stark contrast to EWAS BMI-DMC loci where DNAm variation appears to be primarily a consequence of BMI32.
In summary, this work advances our understanding of the mQTL landscapes across genetic ancestries, shedding light on the underlying mechanistic models that shape this complex landscape and their downstream effects on cellular processes and diseases/phenotypes. The associated mQTL database in EAs constitutes an invaluable resource for future studies to help explain differential susceptibility to disease and complex trait variation across different ancestries. As the SNPs studied here are microarray-based tagged SNPs, it is possible that the true causal SNPs are not captured. In the future, the influence of rare variants on DNAm could also be investigated.
Methods
Further details of experimental methods and data analyses are provided in Supplementary Note.
Participants
Samples in the discovery dataset included 3,523 Han Chinese volunteers recruited in three regional districts of China: Zhengzhou, Taizhou and Nanning (NSPT, 1,310 males and 2,213 females, aged from 18 to 83 years old, mean ± s.d. = 50.21 ± 12.75) from May 2015 to May 2019.
Samples in the validation dataset CAS included 1,060 Han Chinese volunteers recruited from the CAS cohort (CAS, 634 males and 426 females, aged from 22 to 64 years, mean ± s.d. = 40.87 ± 9.41) from Jan 2021 to Jun 2021. In brief, the CAS cohort is a prospective multi-omics cohort that enrolled 3,102 CAS employees (49.0%) from various CAS institutes or offices located in Beijing, China. They were characterized by their high level of education, being in the young- to middle-age range, and having a primary origin from the Chinese Han population. Eligible participants completed a baseline questionnaire regarding lifestyle, medical history and health-related questions, and underwent physical examinations by trained physicians at Beijing Zhongguancun Hospital. Fasting blood samples were collected for clinical laboratory tests and generation of omics data. All participants had genomic data and a subgroup of them (n = 1,071) had deep molecular phenotypic data, including data on epigenetics, proteomics and metabolomics.
Samples in the validation dataset CGZ included 798 Han Chinese volunteers recruited at baseline in two CGZ clinical trails (NCT02121717, also known as CGZ301, and NCT02173457 also known as CGZ302) from 2014 to 2017; a new pan-PPAR agonist developed by Shenzhen Chipscreen Biosciences was used for treating type 2 diabetes51,52,53. The CGZ cohort included 492 males and 306 females (aged from 24 to 70 years, mean ± s.d. = 51.0 ± 9.7).
Ethics
The discovery cohort is a subproject of The National Science and Technology Basic Research Project, which was approved by the Ethics Committee of Human Genetic Resources of the School of Life Sciences, Fudan University, Shanghai (14117). CAS study protocol was approved by the Institutional Review Board of Beijing Institute of Genomics and Zhongguancun Hospital (2020H020, 2021H001 and 20201229). CGZ consists of two registered clinical trials, CGZ301 (NCG02121717, 26 sites) and CGZ302 (NCG02173457, 33 sites). Ethical approvals were obtained from the ethical committees of the 59 study centers. All procedures performed in the study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the Declaration of Helsinki and its later amendments or comparable ethical standards. All participants provided written informed consent.
Statistics and reproducibility
Power estimation indicated that the discovery cohort (NSPT, n = 3,523) has sufficient power to detect mQTLs at MAF of 0.01 when DNAm variance explained is larger than 2% (note that in the study, we found that the median DNAm variance explained by a mQTL was 3.1%, with an interquartile range of 1.9%–6.3%). The two validation cohorts (CAS, n = 1,060 and CGZ, n = 798) have sufficient power to detect mQTLs at MAF of 0.01 when DNAm variance explained is larger than 5% or 6.5%. No statistical method was used to predetermine the sample size. The samples used in this study have already excluded those who failed in the step of quality control (QC) of the SNP chip or DNAm chip. No data were excluded in the following analysis.
DNAm assessment of the discovery panel (NSPT)
Blood samples were kept in Fudan University Taizhou Institute of Health Sciences for storage at −80 °C until DNA extraction. DNA extraction was performed using a TGuide M48 Automated nucleic acid extractor. Genome-wide DNAm was profiled using the Infinium MethylationEPIC BeadChips (Illumina). Five hundred nanograms of genomic DNA from each whole blood sample was bisulfite converted using the EZ DNA Methylation Kit (Zymo Research). BeadChips were processed following the manufacturer guide and protocol for Infinium MethylationEPIC array (Illumina). DNA was hybridized to BeadChips and single base extensions were performed using a Freedom EVO robot (Tecan). BeadChips were subsequently imaged using the iScan Microarray Scanner (Illumina). Illumina.idat files were then processed with the minfi Bioconductor package v1.46.0 (ref. 54) without background correction (although background correction reduces bias, it does so at the expense of increased variance, which is generally something to be avoided, unless the DNAm data are used for copy-number estimation). Probes with SNPs were removed using the dropLociWithSnps function from minfi. This function uses the SNP information provided by Illumina and UCSC Common SNP tables (including versions 132, 135, 137, 138, 141, 142, 144, 146 and 147) with preset MAF (0 is the default value and was used here) to filter SNP CpGs. We further removed probes on chromosomes X and Y. We further used the Illumina definition of β values and derived P values of detection for the rest of the probes by comparing the total intensity U + M to that of the background distribution (given by negative control probes), as implemented in minfi. β values with P values of detection greater than 0.01 were set to NA. Of note, the threshold of detection (P < 0.01) is more stringent than the P < 0.05 threshold used in the other cohorts, partly because sample coverages were very high, allowing for a more stringent threshold while also retaining a high coverage over probes. Only probes with less than 5% missing values were retained. The missing β values were then imputed with the impute.knn function (using k = 5) in R. Type 2 probe bias was corrected using BMIQ55. All this resulted in an 811,876 probes times 3,523 samples data matrix. Based on principal component (PC) analyses, we found a significant slide/beadchip effect. Therefore, we used ComBat56 on M values (logit of β values) to correct for the slide effect and then M values were used in mQTL mapping.
SNP genotyping and imputation of the discovery panel (NSPT)
Genome-wide SNP genotyping was profiled using Illumina Infinium Global Screening Array, which analyzes over 710,000 SNPs. It is a fully custom array designed by WeGene (https://www.wegene.com/). Samples with call rate <98%, ambiguous sex and duplicates were excluded. No sample failed the heterozygosity and inbreeding test. 3,523 samples were kept in discovery NSPT dataset. SNPs were excluded if they had a call rate <98%, MAF < 1% and P value of violations from Hardy–Weinberg equilibrium (HWE) (PHWE) < 0.001. After QC, 433,485 SNPs of 3,523 samples are left for further analyses. Imputation was done by SHAPEIT2 and IMPUTE2 using the 1000 Genomes Phase 3 as a reference. Imputed variants were filtered with MAF > 0.01, imputation quality score >0.8 and violations from HWE (P < 1 × 10−5), leaving 8,615,463 variants. Then, we excluded SNPs that had multiple alleles that belonged to insertion or deletion. After that, there were 7,576,990 SNPs (tested) left for mQTL mapping.
DNAm assessment of the validation panel (CAS)
Blood samples of CAS participants were taken after fasting overnight for at least 8 h. Blood samples were stored at −80 °C freezers in the Beijing Institute of Genomics until DNA extraction. DNA extraction was performed using XPure Blood DNA Extraction Kit (Biokeystone), then the DNA was bisulfite converted using the EZ DNA Methylation Kit (Zymo Research). Genome-wide DNAm was profiled using the Infinium MethylationEPIC BeadChips (Illumina). BeadChips were processed following the manufacturer guide and protocol for the Infinium MethylationEPIC array. BeadChips were subsequently imaged using the iScan Microarray Scanner (Illumina). The resulting Illumina.idat files were processed with the ChAMP Bioconductor package v2.30.0 (ref. 57) and most of the operations followed the recommendations of the package authors and predecessors58. We extracted the methylation β values, checked the consistency of replicates (n = 5) and compared the control genotype probes (59) on the EPIC with our genotype data to make sure there were no mislabeling samples and data quality issues. Then we did some filters on probes and samples based on quality. We removed the samples with missing probes of more than 10% and the probes missing in samples of more than 20%, then carried out KNN imputation for the rest as recommended by the package. We removed the probes with less than three beads in more than 5% of samples and the non-CpG, cross-reactive or nonspecific ones. The probes mapping on the sex chromosomes and contained SNPs (the SNP list provided in ref. 59) were also removed. After checking sample outliers by MDS plots and β value distribution of type 1 and type 2, we did data normalization to convert the type 2 to type 1 probes using BMIQ55, which could reduce the differences between them and improve reproducibility. We kept the samples that had genotype data and transformed the methylation β values into M values. Finally, 751,015 methylation probes and 1,060 samples were left for the following analysis.
SNP genotyping and imputation of the validation panel (CAS)
Genomic DNA was extracted from peripheral blood samples and genotyped on the Infinium Asian Screening Array + MultiDisease-24 (ASA + MD) BeadChip, a specially designed genotyping array for clinical research of EA population with 743,722 variants on it. The GenTrain v2.0 in GenomeStudio was used to perform genotype calling. Individuals with sex mismatch, biological relatedness, possible contamination or departure from the Chinese Han population were removed (n = 0). We regenotyped 15 samples with a low genotyping call rate (<98%). As a result, the mean call rate for all samples was 99.18%. For SNP level QC, variants were excluded if they were duplicates, not on autosomal chromosomes, had a MAF less than 1%, had a missing call rate ≥5% or had a HWE P value less than 1 × 10−4. Imputation of unmeasured SNPs was performed using SHAPEIT2 and IMPUTE2 with the 1000 Genomes phase 3 as a reference panel. SNPs with more than 5% missingness, imputation info score less than 0.6, or MAF less than 1% or that showed significant deviation from HWE were further excluded from the further analysis. After removing samples without DNAm data, 3,455,470 SNPs and 1,060 samples were left for the following analysis.
DNAm assessment of the validation panel (CGZ)
Blood samples of the CGZ cohort were kept in Shenzhen Chipscreen Biosciences Co. Ltd. until DNA extraction. DNA extraction was performed using QIAamp DNA Blood Mini Kit (Qiagen). Genome-wide DNAm was profiled using the Infinium MethylationEPIC BeadChips (Illumina). Five hundred nanograms of genomic DNA from each whole blood sample was bisulfite converted using the EZ DNA Methylation Kit (Zymo Research). BeadChips were processed following the manufacturer guide and protocol for Infinium MethylationEPIC array. DNA was hybridized to BeadChips and single base extension was performed using a Freedom EVO robot (Tecan). BeadChips were subsequently imaged using the iScan Microarray Scanner (Illumina). Illumina.idat files were then processed with the CHAMP Bioconductor package v2.30.0 (ref. 57), which resulted in a 717,100 probes times 798 sample data matrix. β values were transformed into M values and M values were used in mQTL mapping.
SNP genotyping of the validation panel (CGZ)
CGZ samples and QC samples were genotyped with Affymetrix Axiom PMR Arrays under instructions from the manufacturer. The raw genotyping data (CEL files) were generated with the GeneTitan workflow, and subsequently went through the Best Practices Workflow embedded in Axiom Analysis Suite for genotype calling. Each sample was genotyped with 902,560 probes. All samples passed a default Dish QC threshold of 0.82 and a QC call rate threshold of 0.97 and had a call rate over 0.98. After the successful completion of the Best Practices Workflow, genotyping calls in VCF and numeric call codes were exported, as well as the QC tables for samples and probes. After that, 874,438 SNPs were left for mQTL mapping.
mQTL-mapping analysis in NSPT
We adopted a two-step analysis strategy for mQTL mapping. Step 1, we adjusted methylation M values for bisulfite slide number, batch, age, sex, predicted blood cell fractions24, top ten genetic PCs and top two DNAm PCs by linear model regression. Then we tested associations between methylation residuals and SNP dosages using fastQTLmapping60 and retained significant SNP–CpG pairs at a loose threshold (P < 1 × 10−10). Step 2, for the screened mQTLs in Step 1, we excluded the outliers of methylation M values (out the range of mean ± 3 s.d.) and the genotypes that presented in less than five samples, then repeated SNP–CpG association analyses in R. We applied Bonferroni correction to maintain an experiment-wide type 1 error rate of 0.05 for cis, lcis and trans pairs, respectively, that is, Pcis < 0.05/4.7 × 109 = 1.06 × 10−11, Plcis < 0.05/1.75 × 1010 = 2.86 × 10−12 and Ptrans < 0.05/6.13 × 1012 = 8.16 × 10−15. After that, we got the list of study-wide significant cis-, lcis- and trans-mQTL pairs, including 56,289,777 cis-mQTL associations between 5,483,276 SNPs and 267,891 CpGs, 2,270,491 lcis-mQTL associations between 271,994 SNPs and 7,746 CpGs and 4,357,250 trans-mQTL associations between 1,138,024 SNPs and 26,415 CpGs. These mQTL results will be used in subsequent analyses unless otherwise specified. We also applied R package MatrixEQTL v2.3 (ref. 61) in the mQTL-mapping study to cross-validate the mQTL-mapping results.
mQTL replication in CAS
To validate our significant results in CAS, we matched our mQTL SNPs and CpGs in CAS according to genomic positions first. Then, we excluded SNPs with palindromic alleles, A/T and C/G. For the left SNPs, we harmonized (switch and flip) the alleles if needed. After preprocessing, there were 20,917,614 (33.25%) mQTL pairs including 1,969,978 (35.41%) SNPs and 224,990 (79.19%) CpGs that were available in CAS. Then, we performed mQTL mapping by fastQTLmapping in a linear model adjusted for age, sex, bisulfite slide number, bisulfite array position, estimated blood cell fraction (B cells, CD4+ and CD8+ T cells and NK cells and monocytes) and top ten genomic PCs. The P values were adjusted for multiple testing using the Benjamini–Hochberg method to control FDR and 19.62 M (93.81%) were successfully replicated (FDR < 0.05). For these 19.62 M mQTLs, we compare the scaled effect sizes between the two cohorts using\(\,{\beta }_{{ijc}}^{s}\), which is defined as the mQTL effect sizes divided by the maximum of absolute effect values in each cohort.
where \({\beta }_{{ijc}}\) is the effect size of mQTL SNP i on CpG \(j\) in cohort \(c\).
Based on \({\beta }_{{ijc}}^{s}\), we assessed the similarity of effect patterns and directions of mQTLs among the two cohorts. The similarity of the pattern was estimated by Spearman’s rank correlation.
mQTL replication in CGZ
To validate our results, we conducted mQTL analysis using another independent Han Chinese cohort (CGZ). First, we matched our mQTL SNPs and CpGs in CGZ according to genomic positions. Then, we excluded SNPs with palindromic alleles, A/T and C/G. For the left SNPs, we harmonized (switch and flip) the alleles if needed. After preprocessing, there were 2,463,100 (3.91%) mQTL pairs including 246,256 (4.43%) SNPs and 220,804 (77.71%) CpGs, which were available in CGZ. Then, we performed mQTL mapping by MatrixEQTL in a linear model adjusted for age, sex, batch, bisulfite slide number, top ten genomic PCs and predicted blood cell fractions (B cells, CD4+ and CD8+ T cells and NK cells, monocytes and neutrophils). The P values were adjusted for multiple testing using the Benjamini–Hochberg method to control FDR, and 2.14 M mQTLs (87.05%) were successfully replicated (FDR < 0.05). For these 2.14 M mQTLs, we calculated the scaled effect sizes as mentioned before, then assessed the similarity of effect patterns and directions of mQTLs among the two cohorts. The similarity of the pattern was estimated by Spearman’s rank correlation.
EA-specific mQTLs
We downloaded the summary statistics of mQTLs reported in the most recent meta-analysis of 36 European studies from the GoDMC (n = 27,750)8, which is the largest European mQTL research to date. We matched and excluded SNPs with palindromic alleles (that is, A/T and C/G) between NSPT and GoDMC. We harmonized (switch and flip) the alleles if needed. We also matched CpGs between NSPT and GoDMC. Then we focused on the SNP–CpG pairs comprised of the left SNPs (all with MAFs larger than 1% in both cohorts due to genomic QCs) and CpGs. For comparison, we used the same significance threshold (P < 1 × 10−14) to identify mQTLs in NSPT (n = 2.65 million) and GoDMC (n = 3.46 million) here. For the 3.46 million mQTLs in GoDMC, we calculated the replication rate in NSPT for different MAF bins. First, we divided the GoDMC mQTLs into different MAF bins (bin size: 0.05) according to their MAFs in EUR data from the 1000 Genomes phase 3 and calculated the distribution of the GoDMC mQTLs in each MAF bin. Then we also calculated the distribution of the replicated mQTLs in NSPT according to the MAF bin in NSPT. We further calculated the percentage of shared mQTLs among GoDMC mQTLs in each MAF bin cell, that is, the extent of GoDMC mQTLs being also mQTLs in NSPT. Using a similar strategy, we calculated the extent of 2.65 million NSPT mQTLs being also mQTLs in GoDMC in terms of different MAF bins.
We defined EA-specific mQTLs as NSPT-only mQTLs with significance threshold P value < 1 × 10−14 and with P value > 1 × 10−14 in GoDMC. The presentations of EA-specific mQTLs in another two cohorts, CAS and FHS, were also calculated.
Colocalization analysis of EA-specific mQTLs and traits in BBJ GWASs
We applied the SMR (v1.3.1)62 followed by HEIDI (v1.3.1)62 to investigate the same variants that influence both DNAm and traits based on 248 K EA-specific cis-mQTLs and GWAS summary statistics of 230 traits in BBJ (downloaded from https://pheweb.jp/). We identified 152 CpG-trait associations scattered in 44 genomic loci (<1 Mbp), involving 85 CpGs and 33 traits (PSMR < 3.7 × 10−9, corrected by Bonferroni method, and PHEIDI > 0.05). We also compared the colocalization loci with cis-eQTL (PeQTL < 1.0 × 10−5) from HGVD18,19, where only 3 of 44 loci included cis-eQTLs nearby.
Colocalization analysis of cis-/trans-mQTLs in NSPT and GWASs in BBJ
To demonstrate the value of the NSPT mQTLs in studying the epigenetic mechanism behind genetic associations compared with the GoDMC mQTLs, we performed SMR to identify CpGs mediating the genetic associations and HEIDI to distinguish whether the mQTLs and the genetic associations were influenced by shared causal variants on NSPT cis-mQTLs (2.58 M) trans-mQTLs (365 K) and 107 BBJ GWAS summary statistics (107 shared traits between BBJ and UKBB). We identified 394 significant (PSMR < 3.5 × 10−9, corrected by Bonferroni method, and PHEIDI > 0.05) cis-colocalizations in EAs, which included 216 SNP loci (>1 Mbp), 45 traits and 340 independent CpGs (>1 Mbp for each locus–trait pair). Among these, 144 cis-colocalizations (96 SNP loci, 38 traits and 127 independent CpGs) were not identified (PSMR > 0.05) in Europeans (GoDMC mQTLs + UKBB GWASs), that is, EA-specific cis-colocalizations. We identified 854 significant (PSMR < 5.0 × 10−9, corrected by Bonferroni method, and PHEIDI > 0.05) trans-colocalizations in EAs, which included 46 SNP loci (>1 Mbp), 23 traits and 739 independent CpGs (>1 Mbp for each locus–trait pair). Among these, 541 trans-colocalizations (36 SNP loci, 15 traits and 486 independent CpGs) were not identified (PSMR > 0.05) in Europeans (GoDMC mQTLs + UKBB GWASs), that is, EA-specific trans-colocalizations.
Cell-lineage-specific mQTL mapping using CellDMC
The inference of cell-lineage-specific mQTLs was implemented via the CellDMC24 algorithm (EpiDISH R package v2.8). CellDMC identifies interactions between phenotype and cell lineage fraction, thus allowing for the detection of cell-lineage-specific differentially methylated cytosines (DMCTs). In this application, the phenotype is the genotype of the mQTL, and the DMCTs inferred by CellDMC would be a measurement of the cell-lineage-specific nature of the mQTL. On the NSPT cohort, we ran CellDMC with confounders as additional covariates, which included age, sex, batch and array position. Blood cell fractions (B cells, CD4+ and CD8+ T cells and NK cells, monocytes and neutrophils) were estimated using EpiDISH63,64. Before running CellDMC, DNAm data were normalized for slide number using ComBat. The reason for not including slide numbers within CellDMC itself is that there are only eight samples per slide, adjustment of which thus benefits from a Bayesian shrinkage approach as implemented in Combat. CellDMC was run at two resolutions: 6 cell-type resolution (B cells, CD4+ and CD8+ and T cells, NK cells, monocytes, neutrophils) and 2 cell lineage resolution (lymphoid and myeloid lineage). When running CellDMC at the resolution of two lineages, we summed the estimated cell fractions of B cells, CD4+ T cells, CD8+ T cells, and NK cells to give a total lymphocyte fraction, whereas for the myeloid lineage, we summed the fractions of monocytes, neutrophils and eosinophils.
The above procedure was carried out for mQTLs that had previously passed a relaxed significance threshold of P value < 1 × 10−8. This was done for computational feasibility. To verify that running CellDMC on all mQTLs with P value < 1 × 10−8 will not miss many non-mQTLs, which are cell-lineage-specific mQTLs, we ran CellDMC on 1 million random unrelated SNP–CpG pairs (non-mQTLs) to check that the fraction of cell-lineage-specific mQTLs (declared using a relaxed FDR < 0.05 threshold) is very low. Indeed, it is worth noting that the overwhelming majority of the mQTLs at P value < 1 × 10-8 display small effect sizes, which is consistent with two scenarios (large effect size in only one minor cell type or small effect size across many cell types). Thus, any putative lineage-specific mQTLs we could be missing due to our initial screening must have very small effect sizes (even when evaluated in purified samples of the affected cell type) and their biological relevance would be unclear. Using FDR < 0.05, there were 6,865,414 B-cell-specific mQTLs, 10,378,286 CD4+ T-cell-specific mQTLs, 12,374,232 CD8+ T-cell-specific mQTLs, 14,279,365 NK-cell-specific mQTLs, 7,178,916 monocyte-specific mQTLs, 26,390,493 neutrophil-specific mQTLs, 22,744,847 lymphocyte-specific mQTLs and 63,964,415 myeloid-cell-specific mQTLs.
We note that the observed overlap between lymphoid and myeloid mQTLs (fOVL) can be expressed mathematically as the true fraction of lineage-independent mQTLs (fSHARED) times the sensitivity to detect such an mQTL in the myeloid lineage (SEmye) times the sensitivity to detect it in the lymphoid lineage (SElym). Rearranging this, the fraction of mQTLs shared between lymphoid and myeloid lineages (fSHARED) can be estimated as fSHARED = fOVL/(SEmye × SElym).
To confidently estimate fOVL, the CellDMC threshold for calling mQTLs in each lineage was set to an unadjusted P value < 0.05. To understand this, consider, for instance, an mQTL with a CellDMC unadjusted P value in the myeloid and lymphoid lineages of 1 × 10−10 and 0.001, respectively. This mQTL would be declared as myeloid-specific if we had used FDR < 0.05, but would not be myeloid-specific if using an unadjusted P value < 0.05 threshold. Because of the potentially limited sensitivity, it, therefore, makes sense to use the more relaxed threshold and to declare such mQTLs as nonspecific. We also note that because an mQTL with P value < 1 × 10−8 must be an mQTL in at least one cell lineage, in the subsequent CellDMC analysis, we can certainly relax significance thresholds to ensure a reasonably low false negative rate. The observed overlap between lymphoid and myeloid mQTLs (fOVL), expressed as a fraction of the total number of mQTLs, for which CellDMC was run, was 0.16 (16%).
To estimate the sensitivities to detect shared mQTLs in the myeloid and lymphoid lineages, we extended our simulation framework published in ref. 25 to the mQTL context. Briefly, this simulation uses realistic DNAm profiles from >1,000 s monocytes (myeloid) and >200 CD4 T cells (lymphoid) from ref. 65, introducing mQTLs with various effect sizes in the separate cell types before mixing them together using realistic proportions of myeloid and lymphoid proportions in blood. By running CellDMC on these simulated mixtures, we obtained estimates of CellDMC’s sensitivity (SE) to detect shared mQTLs in the myeloid and lymphoid lineages for a range of different effect sizes. As a definition of effect size, we used the difference in mean DNAm divided by the average of the standard deviations. Finally, we then performed a mathematical integration of the observed effect sizes for all mQTLs with P value < 1 × 10−8 with these power estimates, to obtain overall estimates of the sensitivity to detect a shared mQTL in the myeloid (SEmye ~ 0.57) and lymphoid (SElym ~ 0.3) lineages. The lower sensitivity in the lymphoid lineage is because the lymphocyte fraction is much lower and less variable than the myeloid one in whole blood. From this, we then estimated the true fraction of shared mQTLs to be 0.16/(0.3 × 0.57) = 0.93, that is approximately 93% of mQTLs are shared between myeloid and lymphoid lineages.
Identification of mQTL hotspots
For each trans-mQTL, we dropped its associated trans-mCpGs, which were near the most significant CpG, and iterated until the left trans-mCpGs apart from each other >500 Kbp to exclude potential correlation between CpGs. Then we calculated the number of independent trans-mCpGs associated with each trans-mQTL. To identify trans-mQTL hotspots, we first divided whole-genome trans-mQTLs into distinct loci. This began with the assignment of the trans-mQTL with the largest number of independent trans-mCpGs and all its adjacent trans-mQTLs (<1 Mbp) to a locus. Then we repeated this procedure for the remaining trans-mQTLs until all trans-mQTLs were successfully assigned to their respective loci. After that, we selected the index mQTL, which was associated with the largest number of independent trans-mCpGs in each locus. A locus containing an index trans-mQTL that was associated with more than one independent trans-mCpGs was defined as a mQTL hotspot. The following analyses were focused on the 16 mQTL hotspots, which were associated with more than 100 independent trans-mCpGs.
Enrichment of trans-mCpGs in the motifs of corresponding TFs
We used two motif enrichment tools (TFmotifView66 and PWMEnrich (R package v4.30.0)67) to evaluate if the trans-mCpGs associated with the index mQTL in each hotspot were enriched in the corresponding TF motifs. For each index mQTL, we select the TFs near (<1 Mbp) it and got the motifs from JASPAR 2020 database68. The list of human TFs was downloaded from ref. 69. For TFmotifView, we randomly selected more than 10 K CpGs from all trans-wide CpGs (the distance between SNP and CpG >5 Mbp or on different chromosomes) of the index mQTL and ensured they had a consistent distribution of methylation variation (s.d.) with the interested trans-mCpGs. We then tested whether the flanking regions (±100 bp) of the interested trans-mCpGs enriched for the TF motifs compared with the background by one-tailed hypergeometric test. For PWMEnrich, the enrichment analyses were performed by using a log-normal threshold-free approach (comparing the average affinity of the interested sequences to the average affinity of length-matched sequences from the background) with all trans-wide CpGs of the index mQTL as background.
For hotspot H2, which contained the TF FOSL2, we downloaded FOSL2 ChIP–seq binding signals data from ChIP-Atlas database70 (peak-caller MACS2 Q value < 1 × 10−5) and performed enrichment of the trans-mCpGs (flanking regions, ±100 bp) compared with all trans-wide CpGs of the index mQTL (that is, rs4666078) using one-tailed hypergeometric test.
Enrichment of super enhancer in trans-mQTL hotspots
We checked if the interested TFs or DNA binding proteins (DBP) located in trans-mQTL hotspots were enriched with super enhancers as about two-thirds of the interested TFs were enriched with binding sites in remote mCpG regions. Super enhancers have crucial functions in defining cell identity28,29,71. The link between trans-mQTL hotspots and super enhancer would disclose the biological importance of these hotspots. Three super enhancer databases72,73,74 were used in this study. We checked if there was super enhancer for each interested TF/DBP in trans-mQTL hotspot in different tissues or cell lineages. We applied a one-tailed hypergeometric test to evaluate the enrichment of super enhancers in trans-mQTL hotspots.
Downstream effect of trans-mQTLs on diseases/traits
To further understand the impact of TF-mediated trans-mQTL hotspots on diseases/traits, we first matched the mQTL SNPs in trans-mQTL hotspots against GWAS databases (GWAS Catalog and PhenoScanner v2). We used 1 Mbp as the window size to check if any of the mQTL SNPs in the trans-mQTL hotspots matched with GWAS signals. We then compared whether mCpGs in TF-mediated trans-mQTL hotspots were significantly enriched for disease-associated methylation sites compared to randomly selected CpGs as well as EWAS signals. Further, we combined two-sample MR analysis (R package TwoSampleMR v0.5.6) and interaction analysis to resolve the relationship between TF-mediated trans-hotspots and diseases.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Our mQTL database is available for download at https://www.biosino.org/panmqtl/, which incorporates mQTLs not only in EA (NSPT) but also in published European and South Asian data. The database also supports searching and visualization of genomic, functional and downstream disease/trait hits of mQTLs and mCpGs. The statistics of mQTLs in NSPT and CGZ cohort are available for download at NODE https://www.biosino.org/node under accession number OEP002902, or directly accessed at https://www.biosino.org/node/project/detail/OEP002902. The statistics of mQTLs replicated in CAS is available for download at OMIX https://ngdc.cncb.ac.cn/omix under accession number OMIX004116, or directly accessed at https://ngdc.cncb.ac.cn/omix/release/OMIX004116. The individual-level genotype data is not available because of IRB restrictions due to privacy concerns. The individual-level DNAm data can be requested at https://ngdc.cncb.ac.cn/omix/release/OMIX004363 (NSPT), https://ngdc.cncb.ac.cn/omix/release/OMIX004333 (CAS) and https://www.biosino.org/node/project/detail/OEP002902 (CGZ). Requests are normally processed within 1–3 months. Data usage shall be in full compliance with the Regulations on Management of Human Genetic Resources in China. The DNAm dataset in buccal cells is available by submitting data requests to mrclha.enquiries@ucl.ac.uk; see the full policy at http://www.nshd.mrc.ac.uk/data.aspx. Managed access is in place for this 69-year-old NSHD study to ensure that the use of the data is within the bounds of consent given previously by participants, and to safeguard any potential threat to anonymity because the participants are all born in the same week. The mQTL results of the EUR cohort (GoDMC) were downloaded from http://mqtldb.godmc.org.uk/downloads. The mQTL results of the EUR cohort (FHS) were downloaded from https://ftp.ncbi.nlm.nih.gov/eqtl/original_submissions/FHS_meQTLs/ (date: September 14, 2020). The annotation of CpG probes was downloaded from https://zwdzwd.github.io/InfiniumAnnotation (date: November 25, 2019). Significant GWAS results were downloaded from GWAS Catalog (https://www.ebi.ac.uk/gwas/docs/file-downloads, date: December 25, 2020) and significant EWAS results were downloaded from EWAS Atlas (https://ngdc.cncb.ac.cn/ewas/downloads, date: December 25, 2020). The cis-eQTL results in whole blood were downloaded from GTEx V8 database (https://www.gtexportal.org/home/datasets; date: June 17, 2020) and HGVD (http://www.genome.med.kyoto-u.ac.jp/SnpDB/). The human gene information (Ensembl release v104) was downloaded from GENCODE (https://www.gencodegenes.org/human/release_37lift37.html; date: April 26, 2021), the list of human TFs was from http://humantfs.ccbr.utoronto.ca/download.php (date: April 3, 2020), and the list of House-Keeping genes was downloaded from https://www.tau.ac.il/~elieis/HKG/. Motifs information of TFs was obtained from JASPAR 2020 database (http://jaspar.genereg.net/; date: July 2, 2021) and JASPAR 2022 (date: August 22, 2022). ChIP–seq signals of TFs were downloaded from the ChIP-Atlas database (http://chip-atlas.org/; date: June 2, 2021). Other data sources used in this study include BLUEPRINT mQTLs summary statistics (https://ega-archive.org/datasets/EGAD00001005200); Phenoscanner GWAS summary statistics (http://www.phenoscanner.medschl.cam.ac.uk/); Functional genomic regions from the Functional Annotation of Animal Genomes (FAANG) Project (https://www.faang.org); PCHi-C data (https://osf.io/u8tzp); H3K27ac HiChIP data (https://www.ncbi.nlm.nih.gov/geo/, GSE101498); The DNase-seq data for B cells and T cells and the H3K27ac ChIP–seq data of neutrophil cells (https://www.encodeproject.org); GO terms, KEGG pathways, and Reactome pathways were downloaded from the Molecular Signatures Database (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp); and FANTOM5 (https://fantom.gsc.riken.jp/data/). Experimental Factor Ontology (EFO) (https://www.ebi.ac.uk/ols/ontologies/efo). GWASs in BBJ (https://pheweb.jp/); GWASs in UKBB (https://pan.ukbb.broadinstitute.org/); super enhancer databases (http://www.licpathway.net/sedb/; http://www.asntech.org/dbsuper/; http://www.licpathway.net/SEanalysis/); segmented functional regions from GM12878 cell line (http://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=wgEncodeAwgSegmentation); 15 chromatin states (https://egg2.wustl.edu/roadmap/data/byFileType/chromhmmSegmentations/ChmmModels/coreMarks/jointModel/final/).
Code availability
Code for the analysis is available at GitHub (https://github.com/Fun-Gene/fastQTLmapping) and Zenodo (https://doi.org/10.5281/zenodo.8084877)75. Most operations are carried out by R (https://cran.r-project.org/), and the plots are mainly made by ggplot2 v3.4.2 R package (https://cran.r-project.org/web/packages/ggplot2/index.html). mQTL mapping is performed by fastQTLmapping (https://github.com/Fun-Gene/fastQTLmapping) and R package MatrixEQTL v2.3 (https://cran.r-project.org/web/packages/MatrixEQTL/index.html). Heritability is estimated by GCTA (https://yanglab.westlake.edu.cn/software/gcta/). MKL is available at https://software.intel.com/tools/onemkl. GSL is available at http://www.gnu.org/software/gsl/. Annotation of SNP is based on ANNOVAR (https://annovar.openbioinformatics.org/en/latest/, date: 2020.11.2) and annotation of CpG is based on the manufacturer’s manifest files (date: 2020.10.21). Genotype calling is based on GenomeStudio (https://support.illumina.com/array/array_software/genomestudio/downloads.html). Imputation of SNP chip is based on SHAPEIT2 (https://mathgen.stats.ox.ac.uk/genetics_software/shapeit/shapeit.html) and IMPUTE2 (https://mathgen.stats.ox.ac.uk/impute/impute_v2.html). Enrichment analysis of mQTLs is performed by R package clusterProfiler v4.8.1 (https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html). DNAm processing is based on R package minfi Bioconductor package v1.46.0 (https://bioconductor.org/packages/release/bioc/html/minfi.html) and CHAMP Bioconductor package v2.30.0 (https://bioconductor.org/packages/release/bioc/html/ChAMP.html). Cell-type mQTLs are estimated by CellDMC, which is available as part of the EpiDISH v2.8 Bioconductor R package (http://bioconductor.org/packages/devel/EpiDISH. eFORGE is run with the web server at eFORGE2.0 (https://eforge.altiusinstitute.org/). Sharing Effect of cell-type mQTLs is estimated by R package mashr (https://cran.r-project.org/web/packages/mashr/index.html). The GO and KEGG pathway enrichment analyses of mCpGs are conducted using R package missMethyl v1.34.0 (https://bioconductor.org/packages/3.13/bioc/html/missMethyl.html). Genes enrichment for diseases/traits analysis is performed by the R package disgenet2r v0.99.3 (https://www.disgenet.org/disgenet2r) based on the DisGeNET knowledgebase (date: 2021.6.9). The two-sample MR analysis is conducted using the R package TwoSampleMR v0.4.26 (https://mrcieu.github.io/TwoSampleMR/). The HiChIP loops are processed by HiCCUPS and implemented in the Juicer Tools (v0.7.5) with default parameter settings. The influence of SNPs on REs is calculated using the tool OpenCausal (https://github.com/liwenran/OpenCausal). Colocalization is performed by SMR v1.3.1 (https://yanglab.westlake.edu.cn/software/smr/#Download). Enrichment of mQTL CpGs for TF motifs is performed by TFmotifView (http://bardet.u-strasbg.fr/tfmotifview/) and R package PWMEnrich v4.30.0 (https://bioconductor.org/packages/release/bioc/html/PWMEnrich.html). Phenome-wide association analysis is carried out by PheWAS (https://gwas.mrcieu.ac.uk/phewas).
References
Bonder, M. J. et al. Disease variants alter transcription factor levels and methylation of their binding sites. Nat. Genet. 49, 131–138 (2017).
Huan, T. et al. Genome-wide identification of DNA methylation QTLs in whole blood highlights pathways for cardiovascular disease. Nat. Commun. 10, 4267 (2019).
McRae, A. F. et al. Identification of 55,000 replicated DNA methylation QTL. Sci. Rep. 8, 17605 (2018).
van Dongen, J. et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat. Commun. 7, 11115 (2016).
Gaunt, T. R. et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 17, 61 (2016).
Hannon, E. et al. Leveraging DNA-methylation quantitative-trait loci to characterize the relationship between methylomic variation, gene expression, and complex traits. Am. J. Hum. Genet. 103, 654–665 (2018).
Chen, L. et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167, 1398–1414 (2016).
Min, J. L. et al. Genomic and phenotypic insights from an atlas of genetic effects on DNA methylation. Nat. Genet. 53, 1311–1321 (2021).
McClay, J. L. et al. High density methylation QTL analysis in human blood via next-generation sequencing of the methylated genomic DNA fraction. Genome Biol. 16, 291 (2015).
Lemire, M. et al. Long-range epigenetic regulation is conferred by genetic variation located at thousands of independent loci. Nat. Commun. 6, 6326 (2015).
Banovich, N. E. et al. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 10, e1004663 (2014).
Bell, C. G. et al. Obligatory and facilitative allelic variation in the DNA methylome within common disease-associated loci. Nat. Commun. 9, 8 (2018).
Liu, Y. et al. GeMes, clusters of DNA methylation under genetic control, can inform genetic and epigenetic analysis of disease. Am. J. Hum. Genet. 94, 485–495 (2014).
Kassam, I. A.-O. et al. Genome-wide identification of cis DNA methylation quantitative trait loci in three Southeast Asian Populations. Hum. Mol. Genet. 30, 603–618 (2021).
Hawe, J. A.-O. X. et al. Genetic variation influencing DNA methylation provides insights into molecular mechanisms regulating genomic function. Nat. Genet. 54, 18–29 (2022).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019).
Li, M. et al. EWAS Atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 47, D983–D988 (2019).
Higasa, K. et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J. Hum. Genet. 61, 547–553 (2016).
Narahara, M. et al. Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS ONE 9, e100924 (2014).
Akiyama, M. et al. Characterizing rare and low-frequency height-associated variants in the Japanese population. Nat. Commun. 10, 4393 (2019).
Yengo, L. et al. A saturated map of common genetic variants associated with human height. Nature 610, 704–712 (2022).
Wilson, N. K. et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 7, 532–544 (2010).
Hoang, T., Lambert, J. A. & Martin, R. SCL/TAL1 in hematopoiesis and cellular reprogramming. Curr. Top. Dev. Biol. 118, 163–204 (2016).
Zheng, S. C., Breeze, C. E., Beck, S. & Teschendorff, A. E. Identification of differentially methylated cell types in epigenome-wide association studies. Nat. Methods 15, 1059–1066 (2018).
You, C. et al. A cell-type deconvolution meta-analysis of whole blood EWAS reveals lineage-specific smoking-associated DNA methylation changes. Nat. Commun. 11, 4779 (2020).
Teschendorff, A. E., Jing, H., Paul, D. S., Virta, J. & Nordhausen, K. Tensorial blind source separation for improved analysis of multi-omic data. Genome Biol. 19, 76 (2018).
Li, W., Duren, Z., Jiang, R. & Wong, W. H. A method for scoring the cell type-specific impacts of noncoding variants in personal genomes. Proc. Natl Acad. Sci. USA 117, 21364–21372 (2020).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Chen, M. H. et al. Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell 182, 1198–1213 (2020).
Goyama, S., Huang, G., Kurokawa, M. & Mulloy, J. C. Posttranslational modifications of RUNX1 as potential anticancer targets. Oncogene 34, 3483–3492 (2015).
Wahl, S. et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 541, 81–86 (2017).
Dick, K. J. et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet 383, 1990–1998 (2014).
Mendelson, M. M. et al. Association of body mass index with DNA methylation and gene expression in blood cells and relations to cardiometabolic disease: a Mendelian randomization approach. PLoS Med. 14, e1002215 (2017).
Demerath, E. W. et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum. Mol. Genet. 24, 4464–4479 (2015).
Teschendorff, A. E. et al. Correlation of smoking-associated DNA methylation changes in buccal cells with DNA methylation changes in epithelial cancer. JAMA Oncol. 1, 476–485 (2015).
Gurzov, E. N., Stanley, W. J., Brodnicki, T. C. & Thomas, H. E. Protein tyrosine phosphatases: molecular switches in metabolism and diabetes. Trends Endocrinol. Metab. 26, 30–39 (2015).
Rodriguez-Nunez, I. et al. Nod2 and Nod2-regulated microbiota protect BALB/c mice from diet-induced obesity and metabolic dysfunction. Sci. Rep. 7, 548 (2017).
Gurses, S. A. et al. Nod2 protects mice from inflammation and obesity-dependent liver cancer. Sci. Rep. 10, 20519 (2020).
Kreuter, R., Wankell, M., Ahlenstiel, G. & Hebbard, L. The role of obesity in inflammatory bowel disease. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 63–72 (2019).
Hugot, J. P. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603 (2001).
Liu, P. et al. Foxp1 controls brown/beige adipocyte differentiation and thermogenesis through regulating β3-AR desensitization. Nat. Commun. 10, 5070 (2019).
Palmer, C. J. et al. Cdkal1, a type 2 diabetes susceptibility gene, regulates mitochondrial function in adipose tissue. Mol. Metab. 6, 1212–1225 (2017).
Consortium, U. I. G. et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat. Genet. 41, 1330–1334 (2009).
Anderson, C. A. et al. Investigation of Crohn’s disease risk loci in ulcerative colitis further defines their molecular relationship. Gastroenterology 136, 523–529 (2009).
Hachim, M. Y. et al. An integrative phenotype–genotype approach using phenotypic characteristics from the UAE National Diabetes Study identifies HSD17B12 as a candidate gene for obesity and type 2 diabetes. Genes (Basel) 11, 461 (2020).
Moreno-Navarrete, J. M. et al. Heme biosynthetic pathway is functionally linked to adipogenesis via mitochondrial respiratory activity. Obesity (Silver Spring) 25, 1723–1733 (2017).
Cox, B. et al. A co-expression analysis of the pacental transcriptome in association with maternal pre-pregnancy BMI and newborn birth weight. Front. Genet. 10, 354 (2019).
Huang, L. O. et al. Genome-wide discovery of genetic loci that uncouple excess adiposity from its comorbidities. Nat. Metab. 3, 228–243 (2021).
Oliva, M. et al. DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits. Nat. Genet. 55, 112–122 (2023).
DeFronzo, R. A. Chiglitazar: a novel pan-PPAR agonist. Sci. Bull. 66, 1497–1498 (2021).
Ji, L. et al. Efficacy and safety of chiglitazar, a novel peroxisome proliferator-activated receptor pan-agonist, in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, phase 3 trial (CMAP). Sci. Bull. 66, 1571–1580 (2021).
Jia, W. et al. Chiglitazar monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomized, double-blind, phase 3 trial (CMAS). Sci. Bull. 66, 1581–1590 (2021).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Teschendorff, A. E. et al. A β-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29, 189–196 (2013).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Tian, Y. et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 33, 3982–3984 (2017).
Wu, M. C. & Kuan, P. F. A guide to Illumina BeadChip data analysis. Methods Mol. Biol. 1708, 303–330 (2018).
Zhou, W., Laird, P. W. & Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 45, 22–22 (2016).
Gao, X. et al. FastQTLmapping: an ultra-fast package for mQTL-like analysis. Preprint at bioRxiv https://doi.org/10.1101/2021.11.16.468610 (2021).
Shabalin, A. A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358 (2012).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Teschendorff, A. E., Breeze, C. E., Zheng, S. C. & Beck, S. A comparison of reference-based algorithms for correcting cell-type heterogeneity in epigenome-wide association studies. BMC Bioinformatics 18, 105 (2017).
Zheng, S. C. et al. EpiDISH web server: epigenetic dissection of intra-sample-heterogeneity with online GUI. Bioinformatics 36, 1950–1951 (2019).
Liu, Y. et al. Blood monocyte transcriptome and epigenome analyses reveal loci associated with human atherosclerosis. Nat. Commun. 8, 393 (2017).
Leporcq, C. et al. TFmotifView: a webserver for the visualization of transcription factor motifs in genomic regions. Nucleic Acids Res. 48, W208–W217 (2020).
Stojnic, R. & Diez, D. PWMEnrich: PWM enrichment analysis. R package version 4.30.0. https://bioconductor.org/packages/release/bioc/html/PWMEnrich.html (2021).
Fornes, O. et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48, D87–D92 (2020).
Lambert, S. A. et al. The human transcription factors. Cell 172, 650–665 (2018).
Oki, S. et al. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP–seq data. EMBO Rep. 19, e46255 (2018).
Stower, H. Gene expression: super enhancers. Nat. Rev. Genet. 14, 367 (2013).
Jiang, Y. et al. SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res. 47, 235–243 (2019).
Khan, A. & Zhang, X. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 44, D164–D171 (2016).
Qian, F. C. et al. SEanalysis: a web tool for super-enhancer associated regulatory analysis. Nucleic Acids Res. 47, W248–W255 (2019).
Peng, Q. et al. Code for the mQTL analyses in 2023 Nature Genetics (v1.0). Zenodo https://doi.org/10.5281/zenodo.8084877 (2023).
Acknowledgements
This work is supported by the Strategic Priority Research Program of Chinese Academy of Sciences (grant XDB38000000 to S.W., F.L. and P.J.), the National Natural Science Foundation of China (NSFC; 92249302 to S.W. and T.N., 32325013 to S.W., 32370699 and 32170652 to A.E.T., 81930056 to F.L., 32170657 to Y.Z. and L.S., 32200472 to W.L.), CAS Young Team Program for Stable Support of Basic Research (YSBR-077 to S.W.), CAS Interdisciplinary Innovation Team to S.W., CAS Youth Innovation Promotion Association (2020276 to Q.P.), Shanghai Science and Technology Commission Excellent Academic Leaders Program (22XD1424700 to S.W.), the Strategic Priority Research Program of Chinese Academy of Sciences (grant XDC01000000 to F.L.), the National Key Research and Development Project (2018YFC0910403 to S.W. and 2018YFE0201603 to Y.Z. and L.S.), Ministry of Science and Technology of the People’s Republic of China (2015FY111700 to L.J.), Science and Technology Commission of Shanghai Municipality Major Project (2017SHZDZX01 to L.J., S.W., F.L., Y.Z. and L.S.), 111 Project (B13016 to L.J.), CAMS Innovation Fund for Medical Science (2019-I2M-5-066 to L.J. and J.W.), Shanghai Science and Technology Commission Excellent Academic Leaders Program (22XD1424700 to S.W.), Science and Technology Service Network Initiative of Chinese Academy of Sciences (KFJ-STS-QYZD-2021-08-001 and KFJ-STS-ZDTP-079 to F.L.), Naif Arab University for Security Sciences (NAUSS-23-R18 and NAUSS-23-R19 to F.L.), CAS Young Team Program for Stable Support of Basic Research (YSBR-077 to S.W.), CAS Interdisciplinary Innovation Team to S.W., CAS Youth Innovation Promotion Association (2020276 to Q.P.), China Postdoctoral Science Foundation (2021M693274 and BX2021336 to W.L.). We are grateful to S. Beck from University College London, W. H. Wong from Stanford University and C. Wang from Huazhong University of Science and Technology for helpful discussion, C. Relton and J. Min from the University of Bristol for sharing information about SNPs, CpGs and mQTLs in GoDMC, X. Chen from Taizhou Institute of Health Sciences of Fudan University, Y. Fan from Human Phenome Institute of Fudan University and Y. Hu from CAS for providing materials and samples in this study, and X. Cai and Q. Qian from the University of Chinese Academy of Sciences for helping in data preparation.
Author information
Authors and Affiliations
Contributions
S.W., F.L. and A.E.T. designed and drafted the work. Q.P., X.L., W.L., H.J. and A.E.T. performed the statistical analyses and contributed to data interpretation and writing of the paper. J.L., Q.L., C.E.B., G.L. and S.P. contributed to data analysis. X.G. contributed to the program of QTL mapping (fastQTLmapping). J.L., N.Y., J.Q., L.Y. and G.Z. generated the mQTL database. C.Y. and S.D. contributed to preprocessing of methylation and SNP chip data in the discovery cohort. L.J., J.W., J.T. and Z.Y. contributed to the design and acquisition of data in the discovery cohort NSPT. Q.Z., P.J. and C.Z. contributed to the design and acquisition of data in the validation panel CAS. Y.Z., X.L. and L.S. contributed to the design and acquisition of data in the validation panel CGZ. S.G., Y.L., T.N. and B.W. contributed to the design of this work. All the authors revised this work, approved the submitted version, agreed with personal contributions and are responsible for the integrity of the data and the accuracy of the data analysis.
Corresponding authors
Ethics declarations
Competing interests
X. Lu is an employee of Shenzhen Chipscreen Biosciences. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Carmen Marsit, Matthew Sudermann and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 mQTLs enrichment for different functional elements.
a,b, Enrichment of mQTLs (a) and mCpGs (b) in six functional elements: CTCF-enriched elements (CTCF), enhancers (E), promoters (P), promoter flanking regions (PF), regulatory elements (RE), and TF binding sites (TFBS). The y-axis indicates the fold changes (see Methods) and the significance from the one-tailed hypergeometric test is denoted by different symbols on each bar, that is, *, P < 0.05; **, P < 0.01; ***, P < 0.001. c, Enrichment of cis-mQTL pairs (left), lcis-mQTL pairs (middle), and trans-mQTL pairs (right) in all combinations of the six functional categories (that is, CTCF-E, P-E, RE-E, and etc). A one-tailed hypergeometric test is applied. The fold changes are labeled within each box. d, Proportion of SNP-CpG pairs of mQTL within the same TAD. e, Comparison of distance distributions of mQTLs and that of 3D loops.
Extended Data Fig. 2 The cis-colocalization at chr13q14.11 provides epigenetic evidence for the East Asian-specific height-association (rs7335629-height).
a, The East Asian-specific height signal (rs7335629) is in high-linkage with three SNPs in the colocalization locus at chr13q14.11. b, rs7335629 has potential chromatin interaction with one of the CpGs (cg21067652) that colocalized at chr13q14.11. c, Both rs7335629 and cg21067652 are located in regions of high DNase in several blood cell lines. d, Two-sample MR result indicates that cg21067652 is a causal factor for ELF1 RNA expression in CAGE (N = 2,765). Two-tailed MR egger test is applied. The dot and error bar indicate the beta value and s.e., which is SNP effect on CpG (x-axis) and ELF1 expression (y-axis). The blue dotted line indicates the regression line from MR egger test with beta = -9.19, P = 1.34◊10-4. e, Two-sample MR result indicates that cg21067652 is a causal factor for height in BBJ (N = 165,056). Two-tailed MR egger test is applied. The dot and error bar indicate the beta value and s.e., which is the SNP effect on CpG (x-axis) and body height (y-axis). The blue dotted line indicates the regression line from the MR egger test with beta = -0.31, P = 3.73◊10-9.
Extended Data Fig. 3 The enrichment of cis- and trans-colocalizations in EA-specific colocalizations and functional states.
a, Enrichment of trans- vs cis-mQTLs amongst EA-specific colocalizations vs others in NSPT (left), and amongst EA-specific vs EAS-EUR shared colocalizations (right). Trans-, cis- colocalizations in East Asian is carried out based on mQTLs in NSPT (N = 3,523) and 107 GWASs in BBJ (N = ~170,000). Trans-, cis-colocalization in European is carried out based on mQTLs in GoDMC (N = 27,750) and 107 GWAS traits in UKBB (N = ~500,000) which are overlapped with traits in BBJ. b, Enrichment results of cis- vs trans-colocalization loci in functional elements. Left: enrichment of cis- and trans-colocalization loci in functional elements; Middle, enrichment of East Asian-specific and EAS-EUR shared cis-colocalization loci in functional elements; Right, enrichment of East Asian-specific and EAS-EUR shared trans-colocalization loci in functional elements. c, Enrichment of cis- and trans-colocalization loci in chromatin states. Left, enrichment of cis- and trans-colocalization loci in chromatin states; Middle, enrichment of East Asian-specific and EAS-EUR shared cis-colocalization signals in chromatin states; Right, enrichment of East Asian-specific and EAS-EUR shared trans-colocalization signals in chromatin states. Two-tailed Fisher’s exact test is applied. Each point with an error bar indicates log10-scaled odds ratio and its 95% confidence interval.
Extended Data Fig. 4 The relation between the trans-colocalization at chr21q22.2 and blood cell traits and immune diseases.
a, The geographic distribution of rs80109907 allele frequencies in different populations (1000 Genomes Phase 3) by the Geography of Genetic Variants (GGV) browser (https://popgen.uchicago.edu/ggv). b, The PheWAS result of rs80107709 (https://gwas.mrcieu.ac.uk/phewas). c, The colocalization result of chr21q22.2 with other blood cell count and immune-related diseases. SMR test is applied, and the x-axis indicates the beta estimates from original GWAS while the y-axis shows the -log10(P) of the SMR test. d, Two-sample MR results showing that 39 CpGs are causal for 7 traits (several blood cell count and immune-related diseases) at FDR < 0.05. MR IVW test is applied. Red and blue squares indicate positive or negative causal effect of CpG on trait, while the size of the square indicates -log10(P) of the MR IVW test.
Supplementary information
Supplementary Information
Supplementary Protocols, References and Figs. 1–16.
Supplementary Tables 1–23
Supplementary Table 1: The extent of 3.46 million GoDMC mQTLs being also mQTLs in NSPT for different MAF bins. Supplementary Table 2: The extent of 2.65 million NSPT mQTLs being also mQTLs in GoDMC for different MAF bins. Supplementary Table 3: NSPT-only mQTLs (NSPT P < 10−14) defined with different significance thresholds in GoDMC. Supplementary Table 4: The overlapping of population-specific and nonspecific mQTLs with significant SNPs and associations in GWAS Catalog, UKBB and BBJ. Supplementary Table 5: EA-specific mQTLs met with associations and signals in three groups (BBJ-specific, UKBB-specific and shared). Supplementary Table 6: Colocalization results of EA-specific mQTLs and GWAS signals of 230 traits in BBJ. Supplementary Table 7: A total of 144 locus–trait associations (96 loci and 38 traits) identified by cis-colocalization, especially in EAs based on mQTLs in NSPT and traits in BBJ. Supplementary Table 8: A total of 541 locus–trait associations (36 loci and 15 traits) identified by trans-colocalization, especially in EAs based on mQTLs in NSPT and traits in BBJ. Supplementary Table 9: PheWAS result of rs80109907 from public database (https://gwas.mrcieu.ac.uk/phewas/). Supplementary Table 10: Weaker (compared to basophil count) but significant East Asian-specific trans-colocalizations at the locus on chr21q22.2 involving several blood cell counts and immune-related diseases. Supplementary Table 11: A total of 233 CpGs significantly enriched for motifs of 62 TFs (P < 5.3 × 10−5). Supplementary Table 12: Enrichment of 233 CpGs in the binding sites of 13 TFs was validated by blood cell ChIP–seq data. Supplementary Table 13: Cell-lineage-specific mQTLs calculated for random unrelated SNP–CpG pairs. Supplementary Table 14: mQTL hotspots in NSPT. Supplementary Table 15: The 12 (of 16) hotspots index mQTLs or their high LD (r2 > 0.6) trans-mQTLs in GWAS Catalog (P < 5 × 10−8). Supplementary Table 16: mQTL hotspots on each chromosome in NSPT. Supplementary Table 17: Annotation of 16 trans-mQTL hotspots. Supplementary Table 18: Overlap of nearby TF/DBPs related cis-eQTL and trans-mQTLs in each trans-mQTL hotspot. Supplementary Table 19: Overlap of nearby TF/DBPs related cis-eQTL in GTEX V7 (P < 0.05) and trans-mQTLs in each trans-mQTL hotspot. Supplementary Table 20: Enrichment of SNPs with significant OpenCausal scores in trans-mQTL hotspots. Supplementary Table 21: A total of 21 putative causal mCpGs (rs4666078trans-associated) of blood eosinophil count identified by two-sample MR analysis. Supplementary Table 22: Two-sample MR analysis statistics summary of NFKB1 CpGs. Supplementary Table 23: Two-sample MR analysis statistics summary of BMI EWAS CpGs.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Peng, Q., Liu, X., Li, W. et al. Analysis of blood methylation quantitative trait loci in East Asians reveals ancestry-specific impacts on complex traits. Nat Genet (2024). https://doi.org/10.1038/s41588-023-01494-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41588-023-01494-9
