
Abstract
The fall armyworm (FAW) Spodoptera frugiperda is thought to have undergone a rapid ‘west-to-east’ spread since 2016 when it was first identified in western Africa. Between 2018 and 2020, it was recorded from South Asia (SA), Southeast Asia (SEA), East Asia (EA), and Pacific/Australia (PA). Population genomic analyses enabled the understanding of pathways, population sources, and gene flow in this notorious agricultural pest species. Using neutral single nucleotide polymorphic (SNP) DNA markers, we detected genome introgression that suggested most populations in this study were overwhelmingly C- and R-strain hybrids (n = 252/262). SNP and mitochondrial DNA markers identified multiple introductions that were most parsimoniously explained by anthropogenic-assisted spread, i.e., associated with international trade of live/fresh plants and plant products, and involved ‘bridgehead populations’ in countries to enable successful pest establishment in neighbouring countries. Distinct population genomic signatures between Myanmar and China do not support the ‘African origin spread’ nor the ‘Myanmar source population to China’ hypotheses. Significant genetic differentiation between populations from different Australian states supported multiple pathways involving distinct SEA populations. Our study identified Asia as a biosecurity hotspot and a FAW genetic melting pot, and demonstrated the use of genome analysis to disentangle preventable human-assisted pest introductions from unpreventable natural pest spread.
Similar content being viewed by others
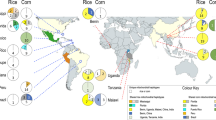
Introduction
Movements of exotic arthropods are increasingly being linked with global agricultural and horticultural trade1,2,3,4,5,6, Europhyte 2018), tourism (e.g.,7), and as hitchhiker pests on aircraft8 and vessels (e.g.,9). These exotic arthropods can have significant negative impact on agricultural production10,11,12,13, and the environments (e.g.,2,14). Good phytosanitary practice together with robust biosecurity preparedness can lower the risk and help mitigate the effects of these invasive species. Invasive arthropods, especially species of agricultural importance, may also introduce novel genotypes (e.g., insecticide resistance genes and alleles;15,16,17) into related native populations through introgression and hybridisation, or into established populations of invasive species, thereby increasing their genomic diversity and further complicating pest management (e.g.,15,18).
The fall armyworm (FAW; Spodoptera frugiperda), a highly polyphagous noctuid moth native to the tropical and subtropical regions of the Americas, was officially reported in Western Africa in early 201619, followed by rapid detections across sub-Saharan Africa (e.g.,20,21,22) by February 2018. In Asia, its occurrence was first reported in South Asia (SA) in India in May 2018, and by December 2018, it had been reported in Bangladesh, Sri Lanka, and in Myanmar (i.e., Southeast Asia, SEA), and in China (i.e., East Asia, EA) by January 2019. In SEA, throughout 2019, detection of FAW was reported from Indonesia23, Laos, Malaysia, Vietnam24, the Philippines25, Cambodia (S Khay pers. ob.), and in EA from the Republic of Korea26,27 and Japan (FAO 2021) in July 2019. The moth's strong flight ability (e.g.,28,29,30) and potential to contaminate certain agricultural trade commodities (e.g.,7) leads to its success as a pest and its detection in increasing number of countries.
In the native range, FAW is conventionally divided into the C-strain and the R-strain31. This distinction is evidenced by behavioural and genotypic differences between the two31. This separation is thought to be an example of incipient speciation and has been primarily seen in North America. However, even in the native range, particularly in the tropics, this distinction seems to break down. Several studies have shown that, at the genetic level, the invasive populations are hybrids (e.g.,32,33,34).
In early 2020, FAW was detected in Pacific/Australia (PA) in Papua New Guinea35,36 and in Australia, FAW was first detected in the Erub and Saibai Islands in the Torres Strait in January 2020, and by early February it was confirmed from maize fields in Queensland (QLD), followed by detection in Western Australia (WA), Northern Territory (NT), New South Wales (NSW), Victoria (VIC), and as of April 2021, it was also reported from Tasmania, as well as Norfolk Island in the Pacific, and most recently (March 2022) also in New Zealand. The FAW is now established across northern Australia and has been reported in a number of crops including sorghum and sugarcane. Since reported by Goergen et al.19, the rapid detections of FAW across the sub-Saharan African nations followed by Asian (i.e., SA, SEA, EA) detections, has led to the general acceptance in the scientific and agricultural sectors that the pest spread eastward from an initial incursion into Africa37. This perception fits the reported patterns of detection, but does it really fit the actual movement of this species across the Old World?
Studies based on nuclear DNA markers involving African (e.g.,33,38) and Asian (e.g.,39) invasive populations identified gene flow patterns and signatures that suggested the FAW’s recent spread was due to multiple introductions, with invasive populations likely to have moved from the east (i.e., EA/SEA) to Africa (e.g.,33). Genomic analyses of Chinese and African FAW populations by Gui et al.40 also detected signatures that were best explained by independent introduction events in EA, and a likely east-to-west movement of the FAW. The authors’ conclusion however, nevertheless maintained the prevailing axiom that FAW arrived in China from western African, with its most recent origin to the Yunnan Province being from the neighbouring country of Myanmar40,41,42,43.
Understanding the spread patterns (e.g., single invasive bridgehead effect44 vs. mass dispersion45) and frequencies of introduction (e.g., single introduction21,40,46,47 vs. multiple introductions32,36,38,39,48,49) of the current invasive FAW populations in Africa, Asia, and Pacific/Australia will have significant implications for the future management of this pest, especially with respect to the delimitation between human-assisted introductions (i.e., preventable via behavioural change) vs. climatic factor-assisted natural migration (i.e., difficult to prevent/not preventable), insecticide resistance management48,49, and climate (i.e., hot and cold) adaptation and tolerance (e.g.50,51,52,53), highlighting most likely agents of spread. It also highlights that evaluating possible future incursion routes makes it necessary to identify weak points in the phytosanitary risk assessments, to enable future targeted monitoring at the right places, and to critically assess official reporting and announcement dates. The implications are significant, such as in reverse simulation studies (e.g.41,42,54,55) that relied on reported detections dates to identify potential wind patterns for explaining pest dispersal patterns, but have not incorporated appropriate genomic/genetic data to ensure consistency between multiple datasets. This could result in significant biosecurity concerns relating to anthropogenic-assisted introductions not being addressed due to the perceived difficulty of preventing pest spread via natural migration.
The aim of this study is to apply genome-wide single nucleotide polymorphic (SNP) loci to: (1) determine the genomic diversity of invasive populations of FAW and the likelihood of multiple introduction events, and (2) determine the area of origin and introduction pathways of FAW populations in Australia. This information can then be used to delineate between human-assisted vs. climate-assisted migration patterns of FAW in SEA and PA regions and inform risk assessments for future incursions of both natural and anthropogenic spread.
Results
Strain assessment
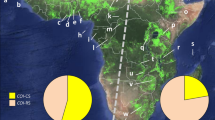
We did not detect any C-strain individual following analysis of 138 fully assembled mitochondrial DNA genomes (mitogenomes) from Australian samples. Our results, particularly that from Northern Territory, are not dissimilar to the finding of Piggott et al.56 who detected only two (i.e., 4.2%) C-strain mtCOI haplotype individuals from a much larger (i.e., n = 48) Northern Territory sample size. Proportions of C-strain to R-strain also varied significantly across the different SEA populations (Table S1) in contrast to the patterns observed in China, India, and African nations (e.g.,22,33,34,39,57). All Australian populations analysed for their corn or rice mitochondrial haplotypes via mitogenome assemblies of whole genome sequencing data therefore contrasted with the invasive populations from SEA where in some countries (e.g., Myanmar, Vietnam) FAW with the C-strain mtCOI haplotypes made up approximately 50% of the populations examined (see Table S1 for C- and R-strains mitogenome proportions, see also Fig. 1 ‘C-strain’ and ‘R-strain’ Maximum Likelihood cladograms).

Maximum Likelihood cladograms of unique Spodoptera frugiperda C-strain and R-strain partial mitochondrial genomes based on concatenation of the 13 PCGs (11,393 bp) using IQ-Tree with 1000 UFBoot replications. Individuals in clades I, II, III, and IV (C-strain) and in Clades I, II, V (R-strain) that are in the same colour scheme (i.e., green, orange, blue, or pinks) shared 100% nucleotide identity. Mitogenome haplotypes from native individuals for both C- and R-strains are in khaki green colour. Red and dark grey dots at branch nodes represent bootstrap values of 87–100% and 74–86%, respectively. Bootstrap values < 74% are not shown. Red arrows indicate invasive individuals’ mitogenome haplotypes that are nested within native individuals. Country and sample codes are listed in Tables S1 and S2.
Mutations in the Triosephosphate isomerase (Tpi) gene used to differentiate the C- and R-strains suggested that virtually all individuals from the SEA, PA, and South Korean (i.e., EA) carried the C-strain genotype at the Tpi gene. Of all SEA and PA individuals assessed, none was homozygous for the rice allele at this locus, while a number of samples (n = 10) were heterozygous C-/R-strains, primarily from the Philippines as well as one sample each from Malaysia and South Korea. Similar to the conclusions of Zhang et al.34, at least in the invasive range, our finding suggested that the Tpi is not a useful marker: not only are the majority of samples of ‘corn’ preference, with limited evidence of hybrids and therefore contrasting greatly with whole genome analysis, but both the Tpi and the mitochondrial markers frequently contradicted each other, and do not provide accurate genealogy in, e.g., hybrid females or their offspring, especially if the female also mated with a hybrid male31.
Mitochondrial genome analysis
In the invasive populations, we identified for the C-strain at least 12 unique maternal lineages forming a total of five clades. For the R-strain, conservatively we identified 19 maternal lineages from nine clades (i.e., I–IX; Fig. 1 ‘C-strain cladogram’). Combining results from this study and the study of Tay et al.33, we therefore identified at least 27 unique maternal lineages in the invasive FAW populations from across 14 countries from Africa, South Asia, East Asia, SEA/Pacific, and Oceania.
The C-strain mitochondrial genomes detected in SEA (Clade II) differed from the one present in the Yunnan CY and Indian populations, although the four haplotypes clustered with high (87–100%) bootstrap support. One other C-strain mitogenome was detected in one individual from China33, and this remained a singleton haplotype amongst the other eight unique C-strain mitogenomes detected from African and SE Asian populations. For the R-strain mitogenomes (Fig. 1 ‘R-strain cladogram’), Clade I consisted of invasive individuals from all countries sampled to-date, although it did not contain individuals from all populations (e.g., no individuals from Australia Sf20-4, Sf20-5, or Mackay populations). R-strain Clade I also contained unique mitogenomes of individuals from the three China Yunnan populations (i.e., CY, XP, YJ), although other Chinese R-strain mitogenome haplotypes were also detected in clades II and VI. The second largest clade (Clade V) contained R-strain mitogenomes shared between individuals from six Australia populations, East Africa (Tanzania, Uganda), India, South Korea, Laos, PNG, and Malaysia (i.e., blue branches), although excluded other populations from SEA (i.e., Myanmar, Vietnam, Philippines), China, and Africa (i.e., Malawi, Benin). Clades II, III, IV, VIII, IX contained mitogenome haplotypes of individuals from either SEA and/or Asia (China, India) but not individuals from any of the African countries. Clades VII, VIII, and IX also have unique mitogenome haplotypes of invasive individuals that were more similar to French Guiana (FG code) or the Caribbean (i.e., Puerto Rico (PR), FG, or Guadeloupe) populations. The overall low bootstrap confidence at various nodes for both C-strain and R-strain phylogenies reflect the high number of maternal lineages with low mitogenome nucleotide diversity (i.e., mitogenome haplotypes differed by only very few nucleotide substitutions) across both invasive and native range populations. These limitations therefore lower their powers for use to confidently infer invasive population origins.
Nuclear SNP phylogenetic analysis
Maximum Likelihood (ML) phylogenetic analysis of the global S. frugiperda populations based on 870 neutral single nucleotide polymorphic (SNP) markers provided evidence of complex genetic relationship especially between individuals representing the invasive populations (Fig. 2). While the New World native populations (USA, Puerto Rico, Guadeloupe, Mexico, French Guiana, Peru, Brazil) clustered together and shared a basal (ancestral) lineage, the invasive populations did not identify Africa as the founding invasive population nor support the axiom of rapid west-to-east spread across the Old World. Selected individuals from Uganda and Benin appeared to cluster close to China/India populations, but there was no strong bootstrap support for a shared common ancestor (i.e., bootstrap value < 74%). China populations (i.e., CY, YJ, XP from Yunnan; see Fig. 1B in58) formed two closely related sister clades with long branch lengths, suggesting these likely represented separate introduction events. Based on analyses of nuclear SNP data, the main Australian FAW population clade consisted of multiple sister clades and were clustered with varying degree of bootstrap support values. Some of these Australian populations appeared to share close relationship with the FE (i.e., South Korean) population (> 87–100% bootstrap support; e.g., selected Burdekin individuals), while others had largely unknown invasive origin(s).

Maximum Likelihood (ML) phylogeny using 879 genome-wide non-coding SNPs with 1000 ultrafast (UF) bootstrap replications. Node support with < 74% are not shown, 74–86% are represented by grey circles, 87–100% are represented by red circles. Country of origin is colour coded.
Southeast Asian countries (i.e., Malaysia, Philippines, Laos PDR, Vietnam, Myanmar) and Pacific/Australia (i.e., PNG) have populations that were mixed, and interestingly were ancestral to East African populations (i.e., Malawi, Uganda), while some Australian populations (i.e., NSW; NT) were shown to be closely related to these SE Asian populations with strong bootstrap support (e.g., > 87–100% support for NT and NSW populations). Myanmar and China appeared to not share FAW populations with similar SNP profiles despite being neighbours. Multiple introductions of FAW with diverse genetic profiles and potentially similarly diverse origins were therefore likely the main reason for their wide spread detection across the Old World.
Clear evidence of multiple introductions can therefore be seen from the phylogenetic analysis, with unique introduction events likely underpinning the establishment of FAW populations in EA. (e.g., China) and SEA (e.g., Malaysia). In Australia, while the NT/NSW populations shared closer evolutionary relationships with SE Asian populations, the population origin for most of Qld and WA populations was unclear, and could be due to a sampling effect (e.g., this study could not source FAW populations from e.g., Indonesia, Cambodia, Thailand, Sri Lanka, Bangladesh, etc.). Some Burdekin and South Korean individuals appeared to share closer evolutionary lineage, although the factor leading to this detected pattern remained as yet unknown.
Population statistics and estimates of substructure
The basic population diversity statistics for each population are listed in Table 1. Nucleotide diversity (π) varied across a narrow range with the lowest (0.237) being from the Malaysian Kedah State population (i.e., reflected its lab-colony background), and the highest from the Malawian population (0.324), similar to that reported (33; π = 0.279–0.329) in native and invasive populations based on the same set of 870 SNP loci. The high nucleotide diversity estimates from the SEA, Australia and South Korean populations likely reflected effects of limited (i.e., 870) highly polymorphic SNPs from non-coding genomic regions being used, and are comparable to the findings from Tay et al.33 that used the same sets of SNP loci. We did not detect significant overall differences between the various invasive populations, and for the SE Asian and Australian populations the nucleotide diversity estimates were generally between 0.258 and 0.291. All invasive populations from Africa (Benin, Malawi, Uganda), SA/EA (India, China (CY, XP, XJ), South Korea), SEA (Malaysia (Johor, Kedah, Penang), Laos, Vietnam, Myanmar, Philippines), and Pacific (PNG)/Australia (WA (Kununurra), NT, QLD (Strathmore, Walkamin, Burdekin), NSW (Wee Waa)) showed higher average observed heterozygosity (Hetobs) than the average expected heterozygosity (Hetexp), with the highest Hetobs seen in the Malawian population as also reported previously33. Within the Australian populations, highest average observed heterozygosity was seen in the Strathmore population, while in populations in SEA (i.e., Myanmar, Philippines) and the EA (i.e., South Korea) all showed similar average Hetobs. Negative average FIS values for all populations were consistent with Hetobs being higher than Hetexp, and suggested populations were out-breeding (i.e., avoidance of matings between related individuals;59). Similar to the previous findings33, the lower Hetexp (i.e., Hetobs > Hetexp; see60) could likewise indicate recent mixing of distinct populations from SEA that suggest multiple introductions (e.g.,33,39 cf.46,47,61,62; i.e., due to a recent bottleneck from a recent western Africa founder event).
The observed heterozygosity excess detected in all invasive range populations could be further explained as due to population sub-structure and isolation breaking through periodic migration. Significant numbers of loci (ca. 30%) were also shown to not be in Hardy–Weinberg equilibrium (HWE) especially for the Malaysian (i.e., Kedah), but also Australian (i.e., Wee Waa, NT, Kununurra), Chinese (e.g., XP), South Korean, and Malawian populations. Taken as a whole, genetic diversity results from this study therefore suggested that the invasive Asian (i.e., SA, SEA, EA) FAW populations exhibited signatures of recent mixing of previously separated populations. Simulated patterns of moth migration of various invasive FAW populations such as between Myanmar and China (e.g.,41,42,55) and to Australia54 are incompatible with the population genomic data, which suggests these were likely discrete and non-panmictic FAW populations with the most probable explanation being due to multiple origins of founding populations.
Genetic differentiation analysis
Estimates of pairwise genetic differentiation (FST) between populations varied significantly (Table 1) and extended to between populations within a country (e.g., Mackay vs. rest of Australia; Kedah vs. rest of Malaysia). Of interest are the pairwise estimates between different Australian FAW populations from Kununurra (Western Australia), Northern Territory, Queensland (Strathmore, Walkamin, Burdekin, Mackay) and New South Wales (Wee Waa) that represented the most recently reported invasive populations in this study, and predominantly showed significant differentiation amongst themselves (with the exception of the two Queensland populations of Mackay and partially for Walkamin) and with other SEA/SA/EA countries. The majority of non-significant population genetic differentiation estimates were in SEA where the presence of FAW was reported earlier, i.e., since 2018 (e.g.,63,64 or as early as 200865,66; see also33), while across Asia (e.g., China) since 2016 but also potentially pre-2014 (16,67; see also33).
Interestingly, significant genetic differentiation was observed between populations from Yunnan province in China and populations from Myanmar, Laos, and Vietnam. Penang and Johor (Malaysia) populations were not significantly differentiated from other SE Asian populations, nor with Ugandan and Malawian populations from east Africa. Individuals from Benin and Mackay (Queensland, Australia) showed non-significant genetic differentiation with all populations except with Kedah, and for Mackay also surprisingly with the Wee Waa population from New South Wales. The South Korean population exhibited significant genetic differentiation with SE Asian population except with Mackay, India and the Yuanjiang (YJ) population in Yunnan Province. Finally, the Kedah population, being one of the earliest collected samples from Malaysia and having been maintained as a laboratory population, showed strong differentiation with all populations (and lowest nucleotide diversity, π = 0.237; Table 2) further supporting unique, non-African, introduction events in SEA. Strong genetic differentiation suggested there was limited gene flow to breakdown sub-structure between populations, and the FST estimates from these invasive populations therefore failed to support a west-to-east spread pathway for the FAW. This observation instead suggested the widespread presence of genetically distinct FAW populations, likely due to independent introductions and therefore also highlighting likely biosecurity weaknesses especially in East Asia (e.g., China, South Korea) and SEA (e.g., Malaysia).
The genetic diversity of Australian populations identified surprisingly complex sub-structure patterns given the short time frame of population detections across different northern Australian regions. Significant genetic differentiation between, e.g., Kununurra (WA), Northern Territory (NT), Queensland (e.g., Strathmore, Burdekin), and Wee Waa (NSW) populations suggests these populations likely derived from separate establishment events. The WA Kununurra population was not significantly differentiated from the Johor State (Malaysia), India and the Cangyuan (CY) China populations, suggesting a potential south-eastern route from SA/SEA into north-western Australia. Contrasting this, Walkamin and Mackay populations showed non-significant genetic differentiation with the Madang (PNG) population, suggesting a potential second pathway for SEA individuals to arrive at the north-eastern region of Australia. Significant genetic differentiation between WA, NT, and Qld populations suggested that at least during the early stage of pest establishment in northern Australia, there was limited gene flow to homogenise the unique genetic background carried by these distinct individuals, some of which exhibited also distinct insecticide resistance profiles48,49.
PCA
We selected specific populations to compare using Principal Component Analysis (PCA) as examples to support evidence of independent introductions, as seen from Fig. 3a between China (CY, YJ, XP) populations vs. Myanmar, in Fig. 3b (within Malaysian populations between those collected from Penang and Johor States vs. Kedah State), in Fig. 3c for between China and East Africa (e.g., Uganda, Malawi), and where Benin and India individuals that grouped with either China or east Africa; and in Fig. 3d between China, Malaysia (Kedah State), and Australia (NT, NSW)). Genetic variability between Australian populations (e.g., Strathmore (QLD) vs. NT and NSW) was also evident (Fig. 3d).

Principal component analysis (PCA) showing variability between selected FAW populations from their invasive ranges. (a) China and Myanmar; (b) Kedah and Johor/Penang populations from Malaysia, (c) China and east African (Uganda/Malawi) populations, (d) Australia (Strathmore, Qld/Northern Territory + New South Wales), China, and Malaysia (Kedah) populations, (e) Australia (Strathmore, Qld) and PNG (Madang Province) populations, (f) Lao PDR/Vietnam and South Korea populations, (g) China and SE Asian (Lao PDR/Vietnam/Myanmar/Philippines/Malaysia) and Pacific/Australia (PNG) populations, and (h) Australia, China and Malaysia (Kedah) populations. Note the overall population genomic variability between countries (e.g., a, c–g) and within countries (e.g., Malaysia (b), Australia (d)). Populations with similar genomic variability are also evident, e.g., for Strathmore (e) and South Korea (f); and for Madang (e) and Lao PDR/Vietnam (f), further supporting potential different population origins of various FAW populations across the current invasive regions. The Southeast Asian and Chinese populations are overall different (g), Australia’s FAW populations showed similarity with both Southeast Asia and China (g, h).
PCA also showed that differences existed between FAW populations from the Madang Province in PNG and with the Strathmore population from Qld (Fig. 3e). The SEA FAW populations from Lao PDR/Vietnam also exhibited diversity from the South Korean population (Fig. 3f), with the South Korean and Strathmore populations largely exhibiting similar diversity patterns, while the Madang population shared similarity with Laos and Vietnam populations. Plotting all SEA populations against China clearly showed that populations from SEA were distinct from the Chinese FAW populations (Fig. 3g), while in Australia, individuals from various populations shared similarity with both Chinese and SEA FAW. Despite the connectedness of the landscape between SEA and China, SEA largely appeared to have their own FAW populations, with FAW in SEA and in China differing in their genome compositions overall as shown via PCA.
PCA further enabled visualisation of genetic diversity amongst Australia FAW populations, suggesting that arrival and establishment of FAW likely involved separate introduction events that followed closely after each other and over a short timeframe. While it had been anticipated that the southward spread of FAW from SEA would necessarily lead to Australia FAW and PNG FAW to share similar genetic backgrounds, the Madang Province FAW population appeared to be different from the Strathmore (Qld) population, with the Madang population being more similar to Lao PDR/Vietnam populations, and the Strathmore population more similar to FAW from South Korea.
DivMigrate analysis
Directionality of gene flow between African, South Asia (Indian), East Asia (China) and SE Asian populations were predominantly from China to east African and SE Asian populations (e.g., Figs. 4a, b, S-1; see also Table 3), while movements of FAW in Laos and Vietnam (i.e., the Indochina region) were predominantly with other SEA countries (e.g., with Myanmar and East Africa; Figs. 4c, d, S-2; see also Table 3) but with no directional movements to the three Yunnan populations (CY, XP, YJ). Migration directionality with other SE Asian populations (e.g., Johor (JB; Fig. S-3) and Penang (PN, Fig. S-4)) showed that these two populations (but especially the Johor population) were predominantly source populations for Uganda, Malawi, Philippines, Vietnam, and PNG (Fig. S-3). Bidirectional migration between Myanmar and Laos PDR populations were also detected with the Johor population from Malaysia (Fig. S-3). When India was selected as the source population, bidirectional migration events were detected with Myanmar and with the Cangyuan (CY) populations (Fig. S-5) while unidirectional migration events from India to Uganda and Malawi and to Laos were detected, and the China Yuanjian (YJ) population showed unidirectional migration to India. Unidirectional migration events from CY and YJ populations to the PNG Madang population were detected, while bidirectional migration events between PNG and Myanmar, Laos PDR, Philippines, Vietnam, and with Uganda and Malawi were also detected (Fig. S-6). No migration events were detected between the West African Benin population and with the South Korean population.

Source populations are CY (a) and XP (b). (c, d) DivMigrate analyses with edge weight setting at 0.453 showing unidirectional (yellow arrow lines) and bidirectional (blue arrow lines) migration between countries in Africa and South Asia/East Asia/SE Asia. Migration rates between populations are as provided in Table 3. (c) Vietnam (VNM) as the source population identified an incidence of unidirectional migration from Malaysia (MYS) Johor state (JB) to Vietnam, while bidirectional migration events were detected from Vietnam to other SE Asian (e.g., Philippines (PHL), Lao PDR (Lao), Myanmar (MMR)), to Pacific/Australia (i.e., Papua New Guinea (PNG)), as well as to east Africa (Uganda (UGA), Malawi (MWI)). (d) Lao PDR (LAO) as source population identified bidirectional migration events between various SEA populations and east African populations, while unidirectional migration events were identified from India (IND) and China (CHN) Yunnan populations (CY, YJ) to Laos PDR. No migration events were evident from SE Asian populations to China.
(a, b) DivMigrate analyses with edge weight setting at 0.453 showing unidirectional (yellow arrow lines) and bidirectional (blue arrow lines) gene flow between countries in Africa and South Asia/East Asia/SE Asia. Significant migration rates (at alpha = 0.5) are in red and as provided in Table 3. Incidences of unidirectional migration were predominantly detected from China (CHN) Yunnan populations (CY, XP) to SE Asian populations (e.g., Myanmar (MMR), Laos PDR (Lao), Philippines (PHL)) and to east African populations (e.g., Uganda (UGA), Malawi (MWI)) (a, b).
Admixture analysis
Admixture analyses involving all Australian, Southeast Asian and South Korean populations from this study; and native populations from the Americas and Caribbean Islands, and invasive populations from Africa (Benin, Uganda, Malawi), India, and China33, provided an overall complex picture of population structure that reflected the species’ likely introduction histories across its invasive ranges.
Admixture analysis that excluded New World, African and Indian populations identified four genetic clusters (i.e., K = 4) to best describe these invasive populations from SEA, and EA (i.e., China, South Korea), and Pacific/Australia (Fig. 5a). At K = 4, Australian populations from NT and NSW, YJ population from China, South Korean, and Malaysia’s Kedah population, each showed unique admixture patterns (i.e., some individuals from NT and NSW populations lacked cluster 3; most of YJ (but also some CY and XP) individuals lacked clusters 1 and 2; South Korean (e.g., MF individuals) lacked cluster 2; Malaysia’s Kedah population lacked evidence of admixture (i.e., reflecting its laboratory culture history) and was made up predominantly by individuals that belonged to cluster 4. Populations from China also differed from most populations from SEA due to the overall absence of genetic cluster 4. Taken as a whole, establishment of the FAW populations in China, Malaysia, vs. other SE Asian populations, and between Australian populations (e.g., NT/NSW cf. WA/Qld), likely involved individuals from diverse genetic background (i.e., multiple introductions). At K = 4, the majority of Australian populations appeared to contain genetic clusters similar to China (i.e., cluster 3) and to SEA (i.e., cluster 2).

Admixture and corresponding CV plots for FAW populations from: (a) Australia, China, South Korea, Lao PDR, Myanmar, Malaysia, Philippines, PNG, and Vietnam, and (b) Benin, China, India, South Korea, Lao PDR, Myanmar, Malaysia, Philippines, PNG, Tanzania, and Vietnam. Optimal ancestral genetic clusters are K = 4 for both admixture plots. Boxed individuals have unique admixture patterns at K = 4 when compared with other populations. China FAW lacked Cluster 2 (navy blue colour; present in almost all SEA and Australian FAW), while in NSW and NT some individuals lacked cluster 3. South Korea ‘MF’ population generally lacked cluster 2, while Kedah (Malaysia) showed distinct (cluster 4) pattern for all individuals. The overall same observations are evident in the admixture plot in (b), with African FAW generally exhibiting admixture patterns similar to SEA populations than to Chinese FAW. With the exception of Kedah (Malaysia) and some Chinese FAW individuals, all FAW in the invasive range showed evidence of genomic admixture (i.e., hybrid signature). The figures were generated using the POPHELPER program < https://pophelpershiny.serve.scilifelab.se/ > and further manipulated in Microsoft PowerPoint for Mac v16.54.
Overall admixture patterns at best K = 4 in China and SEA remained unchanged when analysed together with African and Indian individuals (Fig. 5b; excluded Australia). Benin individuals were either similar to China or to SEA, while eastern African populations (e.g., Uganda, Malawi) were similar to Southeast Asian populations from e.g., Vietnam, Laos, and is in agreement with the phylogenetic inference (Fig. 3) that identified these African individuals as having loci that were derived from Southeast Asian populations.
Genome-wide SNP loci demonstrated that invasive FAW populations from SEA and Australia exhibited admixed genomic signatures similar to that observed in other invasive populations33,34. While the current invasive populations in Africa and Asia likely arrived already as hybrids as suggested by Yainna et al.68, the Malaysia Kedah State population was potentially established by offspring of a non-admixed female. Distinct admixture patterns in Malaysian FAW populations between Kedah and Johor/Penang states therefore suggested that establishment of these populations was likely as separate introduction events. As reported also in Tay et al.33, the Chinese YJ population appeared to have admixed signature that differed from XP and CY populations, and suggested that the YJ population could have a different introduction history than the XP and CY populations. Similar multiple genetic signatures based on lesser nuclear markers by Jiang et al.39 also supported likely multiple introductions of China Yunnan populations.
Discussion
In this study, we demonstrated through genomic analysis of genome-wide SNP markers signatures of multiple independent introductions of S. frugiperda in Asia (i.e., SEA. EA), the detection of distinct population sub-structures between what had been widely assumed (e.g.,41,42,55) as genetically linked populations between Myanmar and China, of multiple introduction pathways into north western and north eastern Australia, and the identification of potential SEA origins of eastern African (i.e., Uganda, Malawian) FAW populations. The findings highlight the need to infer population movements through genome-wide analyses; while critical assessments are needed of conclusions based on movement studies such as reverse trajectory analyses54,55, and inference of population genetics relying solely on detection reports and limited (e.g., partial mitochondrial genes; partial TPI gene) DNA markers. Importantly, the study demonstrated the unexpected complexity of FAW populations across its current sub-Saharan, South Asia, Southeast Asia, East Asia, and Pacific/Australia invasive ranges since the species’ initial 2016 confirmation in western Africa. Results from this study further supported that human (anthropogenic)-assisted activities were likely major contributing factors, as also cautioned by Early et al.7 based on the prevailing opposite wind direction (i.e., east-to-west) in the African continent.
The rapid spread rate of this pest since 2016 to over 70 countries69 has generally been regarded as a result of its long distance migration ability (e.g.,28,29,40,57) following a single introduction into west Africa19,21,46,47,62 and subsequent wind-assisted dispersal37. In contrast, genomic (e.g.,33,38, and genetic39,48) evidence has begun to emerge that the current widespread establishments of FAW populations in Asia were the result of multiple introductions, and likely associated with international agricultural trade. While pre-border interception data from SEA are generally lacking, regular interceptions of FAW in agricultural commodities by Europhyte (e.g., 2018) provided evidence to demonstrate the regular interceptions of FAW from its native countries (and increasingly, also from established invasive populations in Africa) were associated with international trade activities. These ‘bridgehead invasive populations’44 established across Asia and Africa enabled subsequent establishments of localised secondary invasive populations via multiple factors including agricultural trade activities and natural migration (e.g.,7,28,70).
From the analyses presented here, we suggest that the number of independent introductions of FAW is likely underestimated. For example, while our genomic analyses have identified populations in Malaysia, South Korea, and China16,33 with distinct signatures, Jiang et al.39 identified six separate genetic signatures in populations from southern and northern China, and Schlum et al.38 and Tay et al.33 identified a total of at least four separate introductions in African populations (e.g., Kenya, Benin, Uganda, Malawi). Population genetic analyses based on limited partial gene markers have also identified low frequencies of unusual polymorphism patterns in the Tpi and mtDNA gene markers in some African and an Indian FAW (e.g.,47,62,71,72). Similarly, Gui et al.40 also detected signatures suggestive of independent introductions based on genome wide SNP analyses but have nevertheless maintained the African origin theory. While the conclusions by these authors have reinforced the hypothesis of the west–east movement of FAW, increasing whole genome evidence makes it more reasonable to conclude that these earlier single gene findings were also evidence of multiple unrecognised independent introductions.
Our investigation based on previously described genome-wide neutral SNP markers32 enabled integration of the published dataset (i.e., of native New World FAW populations and invasive African, Indian, and Chinese FAW populations) with the current SE Asia, East Asia, and Pacific/Australia FAW populations to reveal a complex picture of the pest’s invasive history. While multiple introductions especially in the Asian continent (e.g., China, Vietnam, Malaysia;16,32,33,39,40,65,66; this study) have played an important role leading to the perception of rapid spread, introductions from elsewhere have also contributed to the African establishment and expansion. Further to genomics evidence, resistance allele characterisation and bioassay experiments have also identified invasive populations with non-overlapping unique insecticide resistance profiles in China (e.g.,73), Indonesia74, Australia48,49, India75, and Africa76,77.
Multiple independent introductions of invasive agricultural pests are well-documented such as for the cryptic whitefly Bemisia tabaci MED and MEAM1 species (e.g.,3,10,78), and for the related noctuid pest Helicoverpa armigera (e.g.,1,5,28), and underpinned the perceived rapid spread (e.g.,79) of these pests in their introduced habitats. Early confirmation of invasive pests in novel habitats is also often challenging especially if initial population sizes were small. For example, while the initial detections of H. armigera in Brazil was during the cropping seasons of 2012/201313,80, its presence in Brazil was only subsequently confirmed to be at least as early as in 200881 and potentially earlier elsewhere (e.g., in Peru) in the South American continent5,82. Similarly for the FAW, its presence in Vietnam was reported since 200865,66,83, and pre-border interceptions of FAW from non-native ranges (e.g., China, Indonesia, Israel, Micronesia, Netherlands, Thailand, and Turkey) back to its native range by the USDA Identification Technology Program (ITP) were pre-201433,67, and pre-dated the 2016 first reported cases of FAW in east Africa19. In Uganda, farmer surveys84,85 showed that FAW damage symptoms on maize crops were first identified in the Namutumba (since 2013) and the Kamuli (since 2014) districts, while Otim et al.86 reported farmers observed FAW damage symptoms in eastern and northern Uganda since 2014, although farmers at the time were unaware of this highly invasive exotic armyworm species. Findings of field surveys84,85,86 were in agreement with anecdotal observations and with single-gene response on Bt maize in South and East Africa, that the FAW could have been present in Africa several years prior to its confirmation in West Africa70.
While the current FAW populations in Vietnam were shown to have a gene flow directionality from Malaysia, whether this could be linked to the 2008 report65,66 of outbreaks around Hanoi remained to be answered. Malaysian populations from Penang and Johor States were also shown to be linked to, and potentially source populations for, Ugandan and Malawian FAW populations (Figs. 2, S3, S4), in addition to the earlier results that also showed Chinese FAW populations to be potential source populations (33; also Fig. 4a, b). Our study and that of Jiang et al.39 therefore identified two potential biosecurity weakness hotspots in Asia that have contributed to the spread of FAW in recent times. The study of Jiang et al.39 as well as the studies of Tay et al.33 and Gui et al.40 therefore identified at least eight unique genetic/genomic signatures of introduction events across different regions (e.g., southern and northern regions) in China. While reverse trajectory studies by, e.g., Wu et al.55 that suggested Yunnan province FAW to have originated from Myanmar, results from the present study showed that there was very little genetic connectivity between them (e.g., Fig. 3a), while DivMigrate analysis showed that gene flow directionality was more likely to be from Cangyuan (CY) to Myanmar (Fig. 4a), thereby highlighting the importance of incorporating genomic analyses to aid interpretations of pest movements.
Southeast Asian and Pacific/Australia countries such as Laos, Myanmar, Vietnam, Philippines, Malaysia, and PNG, appeared to be the geographic ‘genetic melting pot’ for the invasive FAW based on our analyses (Figs. 4a–d; S1–S6), although their population establishment scenarios were different. For example, our analyses suggested that Laos, Myanmar, Philippines, and PNG populations were likely linked to ‘bridgehead invasive founder China’s CY population’, the Vietnam population was more likely associated with individuals from Malaysia’s Johor State ‘bridgehead invasive founder population’ (Fig. 4c). DivMigrate analyses therefore suggested that populations in these SE Asian countries have diverse origins, both as potential direct recipients of New World FAW (e.g., Malaysia, also China) and as recipients of successfully established secondary invasive populations (e.g., Laos, Vietnam, Myanmar), with subsequent bi-directional movements between these countries.
Pest spread into Australia originating from Asia and SEA have been shown to be associated with wind patterns87, and separate introduction pathways for Western Australia and Queensland insect populations (e.g., the blue tongue virus vector Culicoides brevitarsis) have been reported88. Similarly, arrival and subsequent detection of S. frugiperda populations across the northern region of Australia also likely represented separate establishments based on nuclear SNP analysis but did not support the reverse trajectory simulation findings of Qi et al.54. Populations between WA, NT, and Qld were shown to be genetically differentiated, suggesting limited gene flow between these populations at the early stage of invasions. The Kununurra population was found to be less genetically differentiated with the Johor (Malaysia) population (but also the population from India), suggesting a route moving south-east from South Asia/SE Asia into Western Australia, potentially via Indonesia. The PNG population was shown to be not significantly differentiated from the Walkamin (Qld) population, while gene flow directionality analysis linked Yunnan populations (e.g., CY (Fig. 4a) and XP (Fig. 4b)) with the PNG population.
Boaventura et al.74 showed that Indonesian FAW individuals have unique insecticide resistance alleles for the AChE and VGSC genes. In China, Lv et al.73 reported diamide resistance alleles in individuals from Guangxi and Guizhou Provinces. While this study failed to source Indonesian and Cambodian populations (due to the difficulty of sharing and sending biological material from Indonesia and Cambodia to Australia), significant differences in resistance to indoxacarb was nevertheless detected between WA (Kununurra) and Qld (Strathmore) populations48,49, and could be due to the different source populations for the WA and the Qld populations. On-going studies to monitor for potential arrivals of novel resistance alleles and those already reported in Indonesia and China to Australia will therefore be needed to inform and impact future FAW management strategies.
Phylogenetic analyses using both mitochondrial genomes and nuclear SNP loci supported multiple introductions at different scales. At least 12 C-strain mitochondrial genomes based on concatenated PCGs were identified, with some mitochondrial DNA genomes being detected only in SEA populations (e.g., Lao03 in Clade II, Lao12 in clade III). In the R-strain, at least 19 unique mitochondrial DNA genomes were detected (Fig. 1). Interestingly, and despite Australia representing the country with the most recent history of FAW’s incursions in this study, unique Australian mitochondrial DNA genomes not reported elsewhere in our other invasive populations were nevertheless detected (e.g., S3_Sf20-1 (Clade 1), KB1_107_AusRice (Clade III), 03_SF20-5_rice (Clade IV), 52_Sf20-1_Block2_21 (Clade V), and 55_Sf20-1_Block2_24 (Clade IX)). Some of these unique Australian mitogenomes shared close maternal evolutionary lineage with South Korean individuals (e.g., Clade III) that was further supported by nuclear SNP phylogeny (Fig. 2). Whether this represents linked anthropogenic-assisted invasion history remained to be confirmed, although direct flight by adult moths between the two countries would be unlikely due to the large distance (ca. > 7000 km). Regardless, it highlights the challenge faced by biosecurity agencies globally in preventing accidental introductions of alien species.
An important finding from this study has been that the SE Asian populations were largely clustered away from other Asian (i.e., China, Indian) populations, supporting separate establishment histories of S. frugiperda populations in this region, and demonstrating the role played by independent, potentially anthropogenic-assisted introductions. The date of S. frugiperda’s establishment in China and SEA (e.g., Myanmar, Vietnam), though unknown, is potentially as early as pre-2014 (67; see also16,33,65,66) and populations from SEA and China remained largely separate at least as recently as 2019/2020, based on analysis of our 870 SNP loci (see also39 based on microsatellite DNA markers), contradicting the hypothesis of a Myanmar origin for the Yunnan S. frugiperda population41,42,55. Taken as a whole and incorporating population substructure (i.e., FST) analysis, directional migration analysis of invasive S. frugiperda populations identified East Asia (e.g., China, South Korea) and SEA as biosecurity hotspots, while multiple introductions involving genetically distinct S. frugiperda likely occurred in north-western (i.e., Kununurra, WA) and north-eastern (i.e., Burdekin, Qld) Australia.
Population genomic studies via whole genome sequencing have enabled the spread patterns of this invasive pest to be re-evaluated to reveal highly complex scenarios that were likely associated with, or at least initiated by, trade activities. Whole genome sequencing is therefore proposed herein as essential for future biosecurity monitoring, in order to allow population genomics to directly impact on development of management policies and strategies (e.g.,58), and to provide practical information to regulators and to some extent producers on how best to manage new invasive populations. The use of genome-wide SNP loci obtained via whole genome sequencing has enabled the elucidation of pathways to disentangle origins and establishment histories. This, bolsters risk assessment efforts, and provide information to biosecurity agencies and to international inter-governmental agencies to differentiate between natural and human-assisted spread. While it would be unlikely to stop natural migration of pests, accidental introductions via human-assisted spread are preventable. Findings from this study set the precedent that aims to inform biosecurity agencies and policy makers to effect behavioural change in the case where pest establishment and spread were underpinned by poor phytosanitary practices.
Material and methods
FAW specimens for genomic analyses
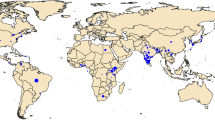
Spodoptera frugiperda populations from the East Asia (i.e., South Korea), Southeast Asia (SEA; i.e., from: Myanmar, Laos, Vietnam, Malaysia, Philippines), and Pacific/Australia (i.e., Papua New Guinea, and Australia (representing new invasive range by S. frugiperda)) were surveyed for whole genome sequencing (Table S1; Fig. 6). Methods of specimen preservation involved collection of larvae/adults from fields were identical and involved placing specimens directly into highly concentrated ethanol (95–99.9%) and storing at − 20 °C until being used in DNA extraction. Samples sent to CSIRO from Queensland (CSIRO code: Sf20-1) and Western Australia (CSIRO code: SF20-4) were first used for bioassays purpose48,49 prior to being stored at -20˚C (once laboratory colonies were established) for DNA extraction purpose. Individuals from these two locations used therefore represent the original field-collected populations from northern Australia.

Countries where invasive populations of FAW surveyed for genomic analyses in this study were collected from (see main text, Table S1). Native New World populations (not shown) and invasive populations from Benin, Uganda, Tanzania, Malawi, India, and China have been reported in Tay et al.33. Geographic regions of the sampled countries were as defined by World Atlas and are provided in Table S1. The map was created by importing the colour-formatted map region from MapChart < https://www.mapchart.net > into Microsoft PowerPoint for Mac v16.54 where location details were added and further manipulated and formatted.
DNA extraction and genome library preparation
DNA of individual FAW samples (Table S1) was extracted using the Qiagen Blood and Tissue DNA extraction kit (Hilden, Germany) and eluted in 200µL elution buffer following the manufacturer’s protocol. Genomic DNA libraries for individual samples were prepared, quantified and sent to the Australian Genome Research Facility (AGRF) in Melbourne (Victoria, Australia) for commercial sequencing.
Processing of genome sequences
Genome sequencing data for each individual were trimmed to remove adapter sequences using Trim Galore! (v 0.6.6; https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and aligned to the S. frugiperda rice genome (v1.0)89 using bwa_mem2 (v2)90. Duplicate alignments were removed using SAMBLASTER (v 0.1.26)91 and sorting completed using SAMtools (v1.9)92). Variants were then predicted using BBMap (v38.90)93 and normalised using bcftools (1.9)92. These variants were then subsampled using 870 SNP’s from Tay et al.33 for further analyses.
Evolutionary and population genomic analyses
For population and evolutionary genomic analyses of the SEA, South Korean, and Pacific/Australian FAW populations, we used the same set of the previously reported 870 neutral genome-wide SNPs33. We also included both native (i.e., Florida, Mississippi, Puerto Rico, Mexico, Guadeloupe, French Guiana, Peru, Brazil) and invasive (i.e., Benin, Malawi, Uganda, Tanzania, India, China) FAW populations that were reported in33 to infer the overall Maximum Likelihood (ML) phylogeny by IQ-Tree94. Based on the phylogenetic analysis, we undertook separate admixture analyses95 by: (1) excluding New World, African, and Indian populations (i.e., include East Asia (China + South Korea) + Southeast Asia + Pacific/Australia), and (2) excluding New World and Australian populations (i.e., including Africa + South Asia (India) + East Asia (Chinese, South Korea) + Southeast Asia + Papua New Guinea).
In the population genomic analysis, native FAW populations were excluded as the implication of multiple introductions that resulted in the establishment of Old World invasive FAW populations (e.g.,32,33,38) meant that inclusion of our limited native populations (i.e., from eight native localities) vs. the higher number of invasive FAW populations (i.e., from 13 invasive countries) were unlikely to provide greater certainty to detect potential origins of invasive populations. The study therefore uses 452 samples with an average sequencing depth of 19.24 (min. 8.71; max. 56.61; median 17.55) for evolutionary genomic interpretations relating to the gene flow patterns of the global FAW populations. Detailed description of the methods has been provided32,33. Admixture was run using multiple K values (3–9), and the cross-validation error (CV) was quantified for each K. The K value with the least CV error (K = 4 in this case) was selected for further interpretation. The 870 SNPs for this present study have been deposited in CSIRO public Data Access Portal96.
Population statistics
We used the populations program in Stacks97 to estimate the observed (Hetobs) and expected (Hetexp) heterozygosities for each population. The number of loci in the global population and individual populations departing significantly from Hardy–Weinberg equilibrium (HWE) was assessed using PLINK 2.098. The populations program in Stacks was used to calculate nucleotide diversity (π) and FIS (99 inbreeding coefficient) and used only variant loci with no window size specified. We used Genepop (v4.7.5)100 to calculate pairwise comparisons of weighted FST values between populations, with significance of differentiation between populations tested using the exact G test.
Mitochondrial Genome (mitogenome) analysis
The mitogenomes of all individuals from SEA (n = 94), East Asia (South Korea, n = 12), and Pacific/Australia (n = 164) were individually assembled following the approach as described in Tay et al.33. Briefly, we used Geneious 11.1.5 to map raw sequences for each FAW individual against a reference genome (GenBank accession number MT897262 (for mtCOI C-strain)/MT897275 (for mtCOI R-strain);33), followed by fine-tuning to resolve regions of poor assembly and ambiguity. Fully assembled mitogenomes were then annotated using MITOS101 by selecting invertebrate mitochondrial genetic code 5. Bed files from MITOS annotations were imported into Geneious 11.1.5 to fine-tune the predicted annotations of gene and protein coding genes (PCGs).
All FAW mitogenomes from this study and those from Tay et al.33 were combined and aligned using MAFFT Alignment v7.450102,103 within Geneious 11.1.5, using the default Auto option for Algorithm, 1.53 for Gap open penalty 0.123 for offset value, and 200 PAM/K = 2 for Scoring matrix. Once aligned, we trimmed and removed all intergenic regions including the A-T rich replication origin, all tRNAs, and the two rRNA genes, leaving 11,303 bp of the 13 concatenated PCGs. We identified unique concatenated PCG mitogenomes within individual countries using the DNAcollapser in FaBox (1.5)104, and partitioned these unique concatenated PCG mitogenomes prior to inferring ML phylogenies without outgroups using IQ-tree implementing UFBoot94 with 1000 replications to ascertain node confidence. Visualisation and presentation of the phylogeny (Fig. 2) was by Dendrogram105 to enable a conservative estimate of the number of maternal lineages in the invasive populations. All assembled and annotated mitogenomes used have been deposited in CSIRO public Data Access Portal106.
Strain assessment
With the non-recombinant nature of the mitogenomes, we analysed the mitogenomes of the FAW according to their strain classification (i.e., C-strain (corn strain) and the R-strain (rice strain)) separately to identify the conservative number of maternal founders for both strains across the invasive ranges, based on concatenation of the 13 PCG regions only, excluding the tRNA and rRNA genes, and the variable intergenic and AT-rich regions. We follow the naming suggestion of Tay et al.31 and refer the rice-strain and corn-strain instead as R-strain and C-strain, respectively. Individual FAWs were characterised as the R- or C- strain by comparing their relevant partial mtCOI gene regions against previously characterised reference sequences (rice (R-strain): GenBank MF197867; corn (C-strain): GenBank MF197868; see22). We note that classification of the FAW host-strain (i.e., R-strain/C-strain) is contentious depending on the markers used (e.g.,107 vs.108) and may have limited applications in hybrid populations31,33, we nevertheless provided this information based on both the partial mtCOI gene identity and the TPI exon 5 gene region as previously described (i.e., for the TPI loci), to differentiate between the C-strain and R-strain S. frugiperda, whole genome sequence data were mapped to the SFRU_RICE_002481 contig89 at location 12939.
Principle component analysis (PCA)
Principal Component Analysis (PCA) was also carried out to increase interpretability of the large and complex FAW genomic dataset through maximising variance by creating new uncorrelated variables and to aid in visualisation. PCA based on the 870 SNPs was performed using PLINK v1.9109 and visualised using ggplot2110. To enable visualisation of evidence of independent introductions we compared (1) China (CY, YJ, XP) populations vs. Myanmar, (2) within Malaysian populations between those collected from Penang and Johor States vs. Kedah State; (3) between China and East Africa (e.g., Uganda, Malawi), and (4) between China, Malaysia (Kedah State), and Australia (NT, NSW)).
DivMigrate analysis
DivMigrate analysis111 was carried out using the complete set of 870 SNP loci for all invasive populations from Africa, Asia, and SEA, with the exception of the Kedah State (Malaysia) population that showed unique genomic background (and represented a population from the on-going lab-maintained culture first collected from maize field in 2019). Australia populations representing the ‘final arrival’ populations, were also excluded in the DivMigrate analysis to increase interpretability relating to East Africa, South Asia, East Asia and SEA populations. Relative migration was assessed using the GST statistics with 100 bootstrap replications and filter threshold (i.e., visualisation of the number of links in the network plot) was set at 0.453 and plotted using the graph visualisation and manipulation software Gephi 0.9.2 < www.gephi.org > and countries were separately drawn using Microsoft PowerPoint for Mac v16.54.
Data availability
All data used in this study have been deposited in the CSIRO Data Access Portal and are freely accessible, under the permanent links https://doi.org/10.25919/24gy-g132 and https://doi.org/10.25919/c53m-ts57.
References
Arnemann, J. A. et al. Multiple incursion pathways for Helicoverpa armigera in Brazil show its genetic diversity spreading in a connected world. Sci. Rep. 9(1), 19380 (2019).
Bedford, G. O. Biology, ecology, and control of palm rhinoceros beetles. Annu. Rev. Entomol. 25, 309–339 (1980).
Elfekih, S. et al. Genome-wide analyses of the Bemisia tabaci species complex reveal contrasting patterns of admixture and complex demographic histories. PLoS ONE 13(1), e0190555 (2018).
Lopes-da-Silva, M. et al. The role of natural and human-mediated pathways for invasive agricultural pests: A historical analysis of cases from Brazil. Agric. Sci. 5, 634–646 (2014).
Tay, W. T. et al. Mitochondrial DNA and trade data support multiple origins of Helicoverpa armigera (Lepidoptera, Noctuidae) in Brazil. Sci. Rep. 7, 45302 (2017).
Tonnang, H. E. Z., Mohamed, S. F., Khamis, F. & Ekesi, S. Identification and risk assessment for worldwide invasion and spread of Tuta absoluta with a focus on sub-saharan Africa: Implications for phytosanitary measures and management. PLoS ONE 10(8), e0135283 (2015).
Early, R. et al. Forecasting the global extent of invasion of the cereal pest Spodoptera frugiperda, the fall armyworm. Neobiota 40, 25–50 (2018).
Porter, J. E. & Hughes, J. H. Insect eggs transported on the outer surface of airplanes. J. Econ. Entomol. 43(4), 555–557 (1950).
Paini, D. R., Mwebaze, P., Kuhnert, P. M. & Kriticos, D. J. Global establishment threat from a major forest pest via international shipping: Lymantria dispar. Sci. Rep. 8(1), 13723 (2018).
De Barro, P. J., Liu, S.-S., Boykin, L. M. & Dinsdale, A. B. Bemisia tabaci: A statement of species status. Annu. Rev. Entomol. 56, 1–19 (2011).
Haile, F., Nowatzki, T. & Storer, N. Overview of pest status, potential risk, and management considerations of Helicoverpa armigera (Lepidoptera: Noctuidae) for US soybean production. J. Integr. Pest Manag. 12(1), 1–10 (2021).
Pozebon, H. et al. Arthropod invasions versus soybean production in Brazil: A review. J. Econ. Entomol. 113(4), 1591–1608 (2020).
Tay, W. T. et al. A brave new world for an old world pest: Helicoverpa armigera (Lepidoptera: Noctuidae) in Brazil. PLoS ONE 8(11), e80134 (2013).
O’Dowd, D. J., Green, P. T. & Lake, P. S. Invasional “meltdown” on an oceanic island. Ecol. Lett. 6(9), 812–817 (2003).
Anderson, C. J. et al. Hybridization and gene flow in the mega-pest lineage of moth. Helicoverpa. Proc. Natl. Acad. Sci. USA 115(19), 5034–5039 (2018).
Tay, W. T. & Gordon, K. H. J. Going global—genomic insights into insect invasions. Curr. Opin. Insect Sci. 31, 123–130 (2019).
Walsh, T. K. et al. Multiple recombination events between two cytochrome P450 loci contribute to global pyrethroid resistance in Helicoverpa armigera. PLoS ONE 13(11), e0197760 (2018).
Valencia-Montoya, W. A. et al. Adaptive introgression across semipermeable species boundaries between local Helicoverpa zea and invasive Helicoverpa armigera moths. Mol. Biol. Evol. 37(9), 2568–2583 (2020).
Goergen, G., Lava-Kumar, P., Sankung, S. B., Togola, A. & Tamò, M. First report of outbreaks of the fall armyworm Spodoptera frugiperda (J E Smith) (Lepidoptera, Noctuidae), a new alien invasive pest in West and Central Africa. PLoS ONE 11(10), e0165632 (2016).
Botha, A. S., Erasmus, A., du Plessis, H. & Van den Berg, J. Efficacy of Bt maize for control of Spodoptera frugiperda (Lepidoptera: Noctuidae) in South Africa. J. Econ. Entomol. 112(3), 1260–1266 (2019).
Nagoshi, R. N. et al. Analysis of strain distribution, migratory potential, and invasion history of fall armyworm populations in northern Sub-Saharan Africa. Sci. Rep. 8, 3710 (2018).
Otim, M. H. et al. Detection of sister-species in invasive populations of the fall armyworm Spodoptera frugiperda (Lepidoptera: Noctuidae) from Uganda. PLoS ONE 13(4), e0194571 (2018).
Trisyono, Y. A., Suputa, S., Aryuwandari, V. E. F. & Hartaman, M. Occurrence of heavy infestation by the fall armyworm Spodoptera frugiperda, a new alien invasive pest, in corn in Lampung, Indonesia. J. Perlindungan Tanaman Indones. 23(1), 156–160 (2019).
Dao, T. H. et al. First record of fall armyworm Spodoptera frugiperda (J.E. Smith), (Lepidoptera: Noctuidae) on maize in Viet Nam. Zootaxa 4772(2), 396–400 (2020).
Navasero, M. V. et al. Detection of the fall armyworm, Spodoptera frugiperda (J.E. Smith) (Lepidoptera: Noctuidae) using larval mrophological characters, and observations on its current local distribution in the Philippines. Philipp. Ent. 33(2), 171–184 (2019).
Kim, J. et al. Development of a simple and accurate molecular tool for Spodoptera frugiperda species identification using LAMP. Pest Manag. Sci. 77(7), 3145–3153 (2021).
Seo, B. Y. et al. The complete mitochondrial genome of the fall armyworm, Spodoptera frugiperda Smith, 1797 (Lepidoptera; Noctuidae), firstly collected in Korea. Mitochondrial DNA B Resour. 4(2), 3918–3920 (2019).
Jones, C. M., Parry, H., Tay, W. T., Reynolds, D. R. & Chapman, J. W. Movement ecology of pest Helicoverpa: Implications for ongoing spread. Annu. Rev. Entomol. 64, 277–295 (2019).
Rose, A. H., Silversides, R. H. & Lindquist, O. H. Migration flight by an aphid, Rhopalosiphum maidis (Hemiptera-Aphididae), and a Noctuid, Spodoptera frugiperda (Lepidoptera-Noctuidae). Can. Entomol. 107(6), 567–576 (1975).
Sparks, A. N. Review of the biology of the fall armyworm (Lepidoptera, Noctuidae). Fla. Entomol. 62(2), 82–87 (1979).
Tay, W. T., Meagher, R. L. Jr., Czepak, C. & Groot, A. T. Spodoptera frugiperda: Ecology, evolution and management options of an invasive species. Annu. Rev. Entomol. 68, 299–317 (2022).
Tay, W. T. et al. Final report on Southeast Asian and Australian FAW population genomics for biosecurity preparedness. Prevention and preparedness for fall armyworm (Spodoptera frugiperda)—Output 2. CSIRO/GRDC 2021, 25 (2021).
Tay, W. T. et al. Global population genomic signature of Spodoptera frugiperda (fall armyworm) supports complex introduction events across the Old World. Commun. Biol. 5(1), 297 (2022).
Zhang, L. et al. Genetic structure and insecticide resistance characteristics of fall armyworm populations invading China. Mol. Ecol. Res. 20(6), 1682–1696 (2020).
Bourke, M. & Sar, S. Fall armyworm in Papua New Guinea: How big is the risk? Australian National University 5 (2020).
Tay, W. T., Kuniata, L., James, W. & Walsh, T. K. Confirmation of Spodoptera frugiperda (Lepidoptera: Noctuidae) in Papua New Guinea by molecular diagnostics of mitochondrial DNA COI gene. BioInvasions Rec. 2022, 125 (2022).
Day, R. et al. Fall armyworm: Impacts and implications for Africa. Outlooks Pest Manag. 28, 196–201 (2017).
Schlum, K. A. et al. Whole genome comparisons reveal panmixia among fall armyworm (Spodoptera frugiperda) from diverse locations. BMC Genom. 22, 179 (2021).
Jiang, Y.-Y., Zhang, Y.-Y., Zhou, X.-Y., Hong, X.-Y. & Chen, L. Population genetics reveal multiple independent invasions of Spodoptera frugiperda (Lepidoptera: Noctuidae) in China. Bull. Entomol. Res. 2022, 1–11 (2022).
Gui, F. R. et al. Genomic and transcriptomic analysis unveils population evolution and development of pesticide resistance in fall armyworm Spodoptera frugiperda. Protein Cell 13, 513–531 (2020).
Li, X. J. et al. Prediction of migratory routes of the invasive fall armyworm in eastern China using a trajectory analytical approach. Pest Manag. Sci. 76(2), 454–463 (2020).
Sun, X.-X. et al. Case study on the first immigration of fall armyworm Spodoptera frugiperda invading into China. J. Integr. Agric. 18, 2–10 (2019).
Zhang, L. et al. Molecular identification of invasive fall armyworm Spodoptera frugiperda in Yunnan Province. Plant Prot. 45(2), 19–24 (2019).
Guillemaud, T., Ciosi, M., Lombaert, E. & Estoup, A. Biological invasions in agricultural settings: Insights from evolutionary biology and population genetics. C. R. Biol. 334(3), 237–246 (2011).
Wilson, J. R. U., Dormontt, E. E., Prentis, P. J., Lowe, A. J. & Richardson, D. M. Something in the way you move: Dispersal pathways affect invasion success. Trends Ecol. Evol. 24(3), 136–144 (2009).
Cock, M. J. W., Beseh, P. K., Buddie, A. G., Cafá, G. & Crozier, J. Molecular methods to detect Spodoptera frugiperda in Ghana, and implications for monitoring the spread of invasive species in developing countries. Sci. Rep. 7(1), 4103 (2017).
Nagoshi, R. N. et al. Genetic characterization of fall armyworm infesting South Africa and India indicate recent introduction from a common source population. PLoS ONE 14(5), e0217755 (2019).
Tay, W. T. et al. Resistance bioassays and allele characterisation inform Spodoptera frugiperda (Lepidoptera: Noctuidae) management and introduction pathways in Asia and Australia. J. Econ. Entomol. 115(6), 1790–1805 (2022).
Tay, W. T., James, B. & Walsh, T. Final report on Australian bioassays. Prevention and preparedness for fall armyworm (Spodoptera frugiperda)—output 2. CSIRO 2021, 31 (2021).
Foster, R. E. & Cherry, R. H. Survival of fall armyworm, Spodoptera-Frugiperda, (Lepidoptera, Noctuidae) exposed to cold temperatures. Florida Entomol. 70(4), 419–422 (1987).
Keosentse, O., Mutamiswa, R., Du Plessis, H. & Nyamukondiwa, C. Developmental stage variation in Spodoptera frugiperda (Lepidoptera: Noctuidae) low temperature tolerance: Implications for overwintering. Austral. Entomol. 60(2), 400–410 (2021).
Yang, X. M. et al. Population occurrence of the fall armyworm, Spodoptera frugiperda (Lepidoptera: Noctuidae), in the winter season of China. J. Integr. Agric. 20(3), 772–782 (2021).
Zhang, D. D., Zhao, S.-Y., Wu, Q.-L., Li, Y.-Y. & Wu, K.-M. Cold hardiness of the invasive fall armyworm, Spodoptera frugiperda in China. J. Integr. Agric. 20(3), 764–771 (2021).
Qi, G. J. et al. Source regions of the first immigration of fall armyworm, spodoptera frugiperda (Lepidoptera: Noctuidae) invading Australia. Insects 12(12), 1104 (2021).
Wu, Q., Jian, Y. & Wu, K. Analysis of migration routes of the fall armyworm Spodoptera frugiperda (J. E. Smith) form Myanmar to China. Plant Prot. 45(2), 1–6 (2019).
Piggott, M. P., Tadle, F. P. J., Patel, S., Gomez, K. C. & Thistleton, B. Corn-strain or rice-strain? Detection of fall armyworm, Spodoptera frugiperda (JE Smith) (Lepidoptera: Noctuidae), in northern Australia. Int. J. Trop. Insect Sci. 41(4), 2607–2615 (2021).
Nagoshi, R. N. et al. Southeastern Asia fall armyworms are closely related to populations in Africa and India, consistent with common origin and recent migration. Sci. Rep. 10(1), 1421 (2020).
Guan, F. et al. Whole-genome sequencing to detect mutations associated with resistance to insecticides and Bt proteins in Spodoptera frugiperda. Insect Sci. 28(3), 627–638 (2021).
Wright, S. The interpretation of population-structure by F-statistics with special regard to systems of mating. Evolution 19(3), 395–420 (1965).
Luikart, G. & Cornuet, J. M. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv. Biol. 12(1), 228–237 (1998).
FAO. Briefing note on FAO actions on fall armyworm in Africa. 2018 (Accessed 13 March 2020); http://www.fao.org/3/a-bt415e.pdf.
Nagoshi, R. N., Goergen, G., Du Plessis, H., van den Berg, J. & Meagher, R. Jr. Genetic comparisons of fall armyworm populations from 11 countries spanning sub-Saharan Africa provide insights into strain composition and migratory behaviors. Sci. Rep. 9(1), 8311 (2019).
FAO. First detection report of the fall armyworm Spodoptera frugiperda (Lepdioptera: Noctuidae) on maize in Myanmar. In 2019, Food and Agriculture Organization of the United Nations, International Plant Protection Convention (2019).
FAO. Fall armywormm (FAW): Impact assessment and the Global Action for Fall Armyworm control 3 (2020).
Nguyen, T. K. O. & Vu, T. P. Checklist of turfgrass insect pests, morphology, biology and population fluctuation of Herpetograma phaeopteralis (Guenee) (Lepidoptera: Pyralidae) in Ha Noi, in spring-summer 2008. In The 3rd National Conference of Ecology and Natural Resources, Hanoi, Vietnam (2009).
Vu, T. P. Insect Pests of Turf Grass, Biology, Ecology and the Control of Herpetogramma phaeoptralis (Guenée) in Hà Nội in Spring Summer 2008 113 (Hà Nội Agriculture University, Vietnam, 2008).
Gilligan, T. M. & Passoa, S. C. LepIntercept, An Identification Resource for Intercepted Lepidoptera larvae. Identification Technology Program (ITP). 2014 (Accessed 4 Dec 2021); http://idtools.org/id/leps/lepintercept/frugiperda.html.
Yainna, S. et al. The evolutionary process of invasion in the fall armyworm (Spodoptera frugiperda). Sci. Rep. 12, 21063 (2022).
Czepak, C. et al. Especial Spodoptera: Migração acelerada. Cultiv. Gd. Cult. 244, 26–29 (2019).
Huesing, J. E. et al. Chapter 01. Integrated Pest Management of Fall Armyworm in AFrica: An Introduction. In Fall armyworm in Africa: A guid or inegrated pest management (eds. Prasanna, B. M. et al.) 120 (CDMX: CIMMYT: Mexico, 2018).
Jing, W. et al. Biology, invasion and management of the agricultural invader: Fall armyworm, Spodoptera frugiperda (Lepidoptera: Noctuidae). J. Integr. Agric. 20(3), 646–663 (2021).
Overton, K. et al. Global crop impacts, yield losses and action thresholds for fall armyworm (Spodoptera frugiperda): A review. Crop Prot. 145, 105641 (2021).
Lv, S. L. et al. Detection of ryanodine receptor target-site mutations in diamide insecticide-resistant Spodoptera frugiperda in China. Insect Sci. 28(3), 639–648 (2021).
Boaventura, D., Martin, M., Pozzebon, A., Mota-Sanchez, D. & Nauen, R. Monitoring of target-site mutations conferring insecticide resistance in Spodoptera frugiperda. Insects 11(8), 545 (2020).
Deshmukh, S. et al. Field efficacy of insecticides for management of invasive fall armyworm, Spodoptera frugiperda (J. E. Smith) (Lepidoptera: Noctuidae) on maize in India. Fla. Entomol. 103(2), 221–227 (2020).
Eriksson, J., Toxicity Bioassays with Insecticide Formulations Used for Control of Spodoptera frugiperda (Lepidoptera: Noctuidae). North West University: South Africa v+86pp (2019).
Worku, M. & Ebabuye, Y. Evaluation of efficacy of insecticides against the fall army worm Spodoptera frugiperda. Indian J. Entomol. 81(1), 13–15 (2019).
de Moraes, L. A. et al. Distribution and phylogenetics of whiteflies and their endosymbiont relationships after the Mediterranean species invasion in Brazil. Sci. Rep. 8, 14589 (2018).
Leite, N. A., Alves-Pereira, A., Corrêa, A. S., Zucchi, M. I. & Omoto, C. Demographics and genetic variability of the New World Bollworm (Helicoverpa zea) and the Old World Bollworm (Helicoverpa armigera) in Brazil. PLoS ONE 9(11), e113286 (2014).
Czepak, C., Albernaz, K. C., Vivan, L. M., Guimarães, H. O. & Carvalhais, T. First reported occurrence of Helicoverpa armigera (Hübner) (Lepidoptera: Noctuidae) in Brazil. Pesq. Agropec. Trop. Goiânia 43(1), 110–113 (2013).
Sosa-Gomez, D. R. et al. Timeline and geographical distribution of Helicoverpa armigera (Hübner) (Lepidoptera, Noctuidae: Heliothinae) in Brazil. Rev. Bras. Entomol. 60(1), 101–104 (2016).
Collins, D., et al. Defra SID5 Research Project Final Report PH0311. In Molecular and morphological identification of the eggs and caterpillars of quarantine listed Noctuidae 2007, Department for Environment, Food and Rural Affairs 21 (2021).
Nguyen, V. D., Ha, Q. H. & Nguyen, T. T. C. Leaves Eating Worm, in Vietnam Insects and Pests, V.L. Pham, Editor (2012).
Kalyebi, A. Documentation of farmer practices that have been useful in Africa for managing the fall armyworm (Spodoptera frugiperda): A case study from Uganda. GRDC 2020, 30 (2020).
Kalyebi, A., Walsh, T. & Tay, W. T. Farmer perception of impacts of Spodoptera frugiperda and transferability of management practices in Uganda. CABI Agric. Biosci. 2022, 125 (2022).
Otim, M.H. et al. The Socio-Economic Impact o Fall Armyworm Invasion on the Production and Income of Maize Value Chain Actors in Uganda. National Agricultural Research Organization, Uganda 64 (2018).
Eagles, D., Daveson, T., Walker, D. P. J., Zalucki, M. P. & Durr, P. Evaluation of long-distance dispersal of Culicoides midges into northern Australia using a migration model. Med. Vet. Entomol. 26(3), 334–340 (2012).
Tay, W. T., Kerr, P. J. & Jermiin, L. S. Population genetic structure and potential incursion pathways of the bluetongue virus vector Culicoides brevitarsis (Diptera: Ceratopogonidae) in Australia. PLoS ONE 11(1), e0146699 (2016).
Gouin, A. et al. Two genomes of highly polyphagous lepidopteran pests (Spodoptera frugiperda, Noctuidae) with different host-plant ranges. Sci. Rep. 7(1), 11816 (2017).
Vasimuddin, M., Misra, S., Li, H & Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In 2019 IEEE 33rd International Parallel and Distributed Processing Symposium (Ipdps 2019) 314–324 (2019).
Faust, G. G. & Hall, I. M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 30(17), 2503–2505 (2014).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25(16), 2078–2079 (2009).
Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner (Lawrence Berkeley National Laboratory, 2014).
Minh, B. Q., Nguyen, M. A. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30(5), 1188–1195 (2013).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19(9), 1655–1664 (2009).
Rane, R. et al. Complex multiple introductions drive fall armyworm invasions into Asia and Australia: Genome-wide single nucleotide polymorphic (SNP) loci of fall armyworm (FAW) Spodoptera frugiperda from Southeast Asia, East Asia, and Pacifi/Australia, in CSIRO Data Access Portal. CSIRO https://doi.org/10.25919/c53m-ts57 (2022).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 22(11), 3124–3140 (2013).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Wright, S. The genetical structure of populations. Ann. Eugen. 15(4), 323–354 (1951).
Raymond, M. & Rousset, F. Genepop (Version-1.2)—population-genetics software for exact tests and ecumenicism. J. Hered. 86(3), 248–249 (1995).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69(2), 313–319 (2013).
Katoh, K., Misawa, K., Kuma, K.-I. & Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14), 3059–3066 (2002).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30(4), 772–780 (2013).
Villesen, P. FaBox: An online toolbox for FASTA sequences. Mol. Ecol. Notes 7(6), 965–968 (2007).
Huson, D. H. & Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 61(6), 1061–1067 (2012).
Rane, R. et al. Complex multiple introductions drive fall armyworm invasions into Asia and Australia: Mitochondrial DNA genomes of Spodoptera frugiperda from Southeast Asia, East Asia, and Australia, in CSIRO Data Access Portal. CSIRO https://doi.org/10.25919/24gy-g132 (2022).
Dumas, P. et al. Phylogenetic molecular species delimitations unravel potential new species in the pest genus Spodoptera Guenee, 1852 (Lepidoptera, Noctuidae). PLoS ONE 10(4), e0122407 (2015).
Nagoshi, R. N. The fall armyworm triose phosphate isomerase (Tpi) gene as a marker of strain identity and interstrain mating. Ann. Entomol. Soc. Am. 103(2), 283–292 (2010).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81(3), 559–575 (2007).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
Sundqvist, L., Keenan, K., Zackrisson, M., Prodöhl, P. & Kleinhans, D. Directional genetic differentiation and relative migration. Ecol. Evol. 6(11), 3461–3475 (2016).
Acknowledgements
We would like to thank Dr Brian Thistleton (Department of Industry, Tourism and Trade, NT), Dr Ian Newton and Dr Melina Miles (QDAF), Mr Jack Daniel (Northern Australia Crop Research Alliance Pty Ltd, Kununurra WA), Prof. Myron Zalucki (University of Queensland), Mrs Anna Madden (Wee Waa, NSW), and Dr Simone Heimoana (CSIRO), for providing Australian FAW specimens for genomic analyses. The Erub Island FAW was kindly provided by the Department of Agriculture, Water and the Environment (DAWE) and Northern Australia Quarantine Strategy (NAQS). Van NTH and Huyen NPT (PPRI, Vietnam) helped with FAW specimen and related metadata collection. DR. Donald Kachigamba provided samples from Malawi. Bill James (CSIRO) helped with laboratory work, Dr Sharon Downes, Bill James, and Dr Sarina Macfadyen (CSIRO), and Dr Eric Huttner (ACIAR) provided helpful discussion during the course of this work. The work performed at INRAE was publicly funded through the ANR (the French National Research Agency, Grant ID 1702-018, given to K.N.) under the “Investissements d’avenir” programme with the reference ANR-10-LABX-001-01 Labex Agro and coordinated by Agropolis Fondation under the frame of I-SITE MUSE (ANR-16-IDEX-0006). It was also funded by a grant from the department of Santé des Plantes et Environnement at Institut national de la recherche agronomique for K.N. (adaptivesv), and was also financially supported by EUPHRESCO (FAW-spedcom, given to Anne-Nathalie Volkoff). The project led by CSIRO is a co-investment by the Grains Research and Development Corporation (GRDC) (CSP2003-008RTX), the Australian Centre for International Agricultural Research (ACIAR) (CROP/2020/144), the Cotton Research and Development Corporation, FMC Australasia and Corteva Agriscience. GRDC manages the project on behalf of the co-investors.
Author information
Authors and Affiliations
Contributions
R.R. data analysis, manuscript preparation. T.K.W. project conceptualisation, manuscript preparation, data analysis. P.L. manuscript preparation, data analysis. A.G. sample preparation, manuscript preparation. T.H.D. project conceptualisation, sample preparation, manuscript preparation. V.L.N. project conceptualisation, sample preparation, manuscript preparation Khin project conceptualisation, T.N.K. sample preparation, manuscript preparation. D.A. project conceptualisation, sample preparation, manuscript preparation. K.C. project conceptualisation, sample preparation, manuscript preparation. M.F. project conceptualisation, sample preparation, manuscript preparation. S.A. project conceptualisation, sample preparation, manuscript preparation. S.S.T. project conceptualisation, sample preparation, manuscript preparation. Y.A.T. project conceptualisation, sample preparation, manuscript preparation. S.K. project conceptualisation, sample preparation, manuscript preparation. J.K. sample preparation, manuscript preparation. L.K. sample preparation, manuscript preparation. K.P. sample preparation, manuscript preparation. A.K. project conceptualisation, manuscript preparation Otim. M.H.O. project conceptualisation, manuscript preparation. K.N. project conceptualisation, sample preparation d’Alençon. Ed'.A. project conceptualisation, sample preparation Gordon. K.H.J. project conceptualisation, data analysis, manuscript preparation. W.T. Tay project conceptualisation, data analysis, manuscript preparation, sourced funding. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rane, R., Walsh, T.K., Lenancker, P. et al. Complex multiple introductions drive fall armyworm invasions into Asia and Australia. Sci Rep 13, 660 (2023). https://doi.org/10.1038/s41598-023-27501-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-27501-x
This article is cited by
-
Strategic Analysis of Collaborative Networks in Spodoptera frugiperda (Lepidoptera: Noctuidae) Research for Improved Pest Management Strategies
Neotropical Entomology (2024)
-
Farmer perception of impacts of fall armyworm (Spodoptera frugiperda J.E. Smith) and transferability of its management practices in Uganda
CABI Agriculture and Bioscience (2023)
-
Biological control of fall armyworm Spodoptera frugiperda (JE Smith) using egg parasitoids, Trichogramma species (Hymenoptera: Trichogrammatidae): a review
Egyptian Journal of Biological Pest Control (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
