
Abstract
The northern giant hornet Vespa mandarinia (NGH) is a voracious predator of other insect species, including honey bees. NGH’s native range spans subtropical and temperate regions across much of east and southeast Asia and, in 2019, exotic populations of the species were discovered in North America. Despite this broad range and invasive potential, investigation of the population genomic structure of NGH across its native and introduced ranges has thus far been limited to a small number of mitochondrial samples. Here, we present analyses of genomic data from NGH individuals collected across the species’ native range and from exotic individuals collected in North America. We provide the first survey of whole-genome population variation for any hornet species, covering this species’ native and invasive ranges, and in doing so confirm likely origins in Japan and South Korea for the two introductions. We additionally show that, while this introduced population exhibited strongly elevated levels of inbreeding, these signatures of inbreeding are also present in some long-standing native populations, which may indicate that inbreeding depression alone is insufficient to prevent the persistence of NGH populations. As well as highlighting the importance of ongoing monitoring and eradication efforts to limit the spread of this species outside of its natural range, our data will serve as a foundational database for future genomic studies into introduced hornet populations.
Similar content being viewed by others

Introduction
The northern giant hornet (NGH) Vespa mandarinia Smith 1852 is widespread across much of southern and eastern Asia, with a distribution ranging from Hokkaido, Japan west to northern India and from far eastern Russia south to Hong Kong1,2. Like other hornets, this species forms annual eusocial colonies and predates upon other arthropods to provision developing larvae3,4. NGH is one of the largest two hornet species and is among the most important predators of honey bees across its endemic range4,5. The sympatric honey bee species Apis cerana possesses natural defenses against NGH, including specialised alarm signals6,7, collection and use of deterrent odorants8, and coordinated thermal defence (‘heat-balling’) against hornet attacks9. Because its natural range does not overlap with that of NGH, the western honey bee Apis mellifera lacks these specialised natural defences6,10 and, consequently, is highly vulnerable to attacks by this predator5,11.
Like other social insects, hornets have a high potential to become invasive4,12,13. At least five hornet species have successfully established populations outside of their native range4: Vespa velutina in Korea, Japan, and much of western Europe; V. orientalis in southern Spain and Gibraltar; V. tropica in Guam; V. bicolor in Taiwan and Spain; and V. crabro in eastern North America. However, genomic data are lacking for the majority of these invasive populations, especially at the earliest stages of introduction. To our knowledge, the only exceptions to this knowledge gap are two studies respectively using V. mandarinia14 and V. velutina15, both of which were restricted to a small number of samples and genetic loci. Better understanding the genetic correlates of these hornet introductions may provide insights into the likelihood that a newly introduced hornet population will become established4. In particular, because of the negative fitness effects of inbreeding depression, the amount of genetic diversity present in an introduced population may be a significant factor limiting the invasive potential of that population16,17. Factors such as polyandry and multiple founding, which increase the genetic diversity present in a single founding event, are therefore likely to contribute to invasion potential in hornets4,15.
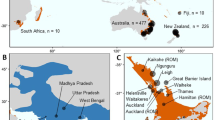
In September 2019, an NGH nest was discovered and eradicated in Nanaimo, British Columbia, Canada14,18. In December of the same year, an NGH specimen was identified in Blaine, Washington, USA, approximately 95 km southeast of the nest found in Nanaimo14. The identification of these samples, together with the known vulnerability of agriculturally important A. mellifera honey bee nests to attacks by NGH, resulted in a concerted effort by agricultural agencies to track and eradicate this hornet species and plan for future introductions. Over the following two years, survey efforts by local government agencies and the public resulted in another 45 confirmed sightings and specimens in British Columbia and Washington, leading to the eradication of four colonies in Washington State (one in 2020 and three in 2021; Fig. 1a–e)19.
No further NGH were detected in Washington or mainland British Columbia in 2022, 2023, or at time of writing in 2024, nor were there any additional confirmed sightings in Nanaimo following the 2019 eradication. These results indicate that some combination of human eradication efforts, Allee effects20,21, ecological factors and chance have likely extirpated the incipient North American NGH populations identified in 2019. Nonetheless, hornets possess a high capacity for invasiveness4, and species distribution models indicate that much of North America may be climatically suitable for the persistence of NGH populations, including areas important for commercial honey production22,23,24. Moreover, analysis of mitochondrial data from two of the samples collected in 2019 indicated separate geographic origins for the Nanaimo and Washington introductions despite the geographic and temporal proximity of the samples14, indicating repeated introductions of the same invasive species within a short window of time. However, that study was based on mitochondrial data alone and included only a small number of individuals (whole mitogenomes for five samples and partial mitochondrial barcode sequences for an additional eight). Those data are therefore insufficient to precisely assess the degree of diversity within the surveyed populations, to estimate the degree to which those populations display signatures of inbreeding, or to address the precise localities from which the invasive introductions originated. A more thorough understanding of the route(s) by which NGH reached North America in 2019 and of the mechanisms that might have limited its spread requires a larger study using more comprehensive genomic data.
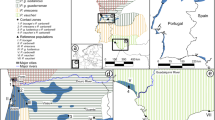
Here, we present the first whole-genome population genomic data from NGH samples collected in the introduced North American range and for samples from native populations in Japan, mainland China, South Korea, and Taiwan (Figs. 1 and 2). These data were sequenced with several objectives in mind: (1) to produce a much higher-resolution survey of the population genetic structure of NGH’s native range than has previously been available; (2) to confirm that the two introduced North American NGH populations have separate origins; (3) to more precisely define the locations of these origins; and (4) to characterize the degree of genomic inbreeding experienced by native and introduced NGH populations, and thereby to determine whether inbreeding depression is likely to present a major obstacle to the establishment of invasive populations of this species.
Using newly sequenced data from 114 NGH samples spanning 7 geographical regions, we generate by far the most comprehensive population genomic survey to date of any hornet species. We confirm that the samples collected in Nanaimo likely had a Japanese origin, while the origin of the Washington introduction is less clear but may be located in South Korea. We further show that samples from the Washington population exhibited strongly elevated levels of inbreeding. However, we also detect inbreeding in a number of native samples, possibly indicating that inbreeding depression alone is insufficient to prevent the persistence of NGH populations. Our results therefore emphasize the necessity of control and eradication efforts to prevent the spread of invasive populations of this species.
Results
Sample collection, read alignment, and variant calling
We sequenced whole genome data for 114 samples from 7 geographical regions (hereafter referred to as populations; Table 1; Fig. 2). Per-sample mean±SE mapping rate of reads was 95.8±0.004%. We mapped a total of 4.4B trimmed reads to 114 samples, with an insert size of 292.6±3.8bp, mean mapping quality of 36.7±0.2 (corresponding to an incorrect base call rate of \(\sim\)0.021%), and an average depth of 19.5±0.4, well above the value required for high-quality SNP calls25. After removing duplicates, 4.3B reads with an average insert size of 292.6±3.7bp and mapping quality 36.6±0.2 remained, with a mean depth of 18.8±0.4 for use in analyses.
After genotyping, the all-sites VCF included 190.5M sites, including 6.01M SNPs across all combined samples. Following filtering for depth and quality, 5.6M SNPs remained across all samples. Numbers of SNPs within each subpopulation before and after pruning for linkage disequilibrium are given in Supplementary Table S1.
For the mitochondrial super-barcoding, after QC and trimming, we analysed an alignment containing 8567 nucleotides across 103 individuals (including reference OQ836204.1 and 102 samples sequenced in this study). Five samples from Fujian, China and one from Japan were removed due to the high proportion of Ns in these sequences. To test a Chinese origin for North American NGH, the reference NGH mitogenome from Yunnan was retained in the alignment for analyses. The resulting alignment contained 422 parsimony informative sites, 42 singleton sites, and 8103 constant sites. The presence of singleton sites mainly resulted from including the single NGH reference sequence from Yunnan, China. From the 103 samples, 30 total mitochondrial haplotypes (mitotypes) were identified with two mitotypes for the five samples from Yunnan, one mitotype for the two Nanaimo samples, seven mitotypes from the 43 South Korean samples (one of which was also shared by all Washington samples), 14 mitotypes from the 14 Japanese samples, five mitotypes from the 13 Taiwanese samples, and a final remaining mitotype for reference sequence OQ836204.1 (Supplementary Table S2).
Confirmation of separate origins for the Nanaimo and Washington introduced populations
Initial PCA using 1.03M LD-filtered SNPs grouped all samples into one of three distinct clusters along PCs 1 and 2, which together captured \(\sim\)25% of variation among samples (Supplementary Figure S1a). Two of these three clusters corresponded to the two mainland Chinese populations (Fujian and Yunnan), while the third included all other samples. Re-running the PCA after excluding the mainland Chinese populations allowed us to more clearly elucidate the variation among the remaining samples. These remaining samples formed four discrete clusters along PCs 1 and 2 (\(\sim\)18% of total variation), each representing a different population, except for a single cluster containing the Japanese and Nanaimo samples (Supplementary Figure S1b). These results held when excluding close relatives (Supplementary Figure S1c,d). PCA therefore strongly supports the possibility that the Nanaimo samples originated from Japan but does not provide the same level of support for a South Korean origin of the Washington samples.
Constructing a maximum-likelihood tree of distantly related individuals using SNP data supported these conclusions (Fig. 3). Most samples were separated strongly by geography in this tree, as expected given the results of the PCA, with samples from the two mainland Chinese populations and Taiwan exhibiting particularly long branches, and the Nanaimo and Japanese samples unequivocally related to one another. The South Korean samples, however, exhibited a more dispersed distribution across the tree, suggesting significant heterogeneity in this region. The Japanese and Washington samples were both nested among the Korean samples on this tree, but this was not the case when considering the TCS network and ML tree generated using mitogenomes alone, which exhibited good separation between Japanese and South Korean mitotypes (Supplementary Figure S2). All 24 samples from Washington state possessed a single mitochondrial haplotype (Vman-6), shared with 14 samples from South Korea. The two samples from Nanaimo, British Columbia possessed mitotype Vman-21. While the Nanaimo samples did not match any Japanese mitotypes exactly, they were nested in a Japanese clade with strong support in the BI and ML analyses and were different from Vman-17 by only three informative SNVs (Supplementary Figure S2). Internal branching support in both ML and BI was low in both methods and reflected in different branching orders (Supplementary Figure S2). However, in both phylogenetic trees and across all methods for quantifying branch support, South Korean, Japanese, and Taiwanese clades were resolved with high support in all cases, including the network analyses, as assessed by the number of informative sites separating the different lineages.
Using a larger SNP-based maximum-likelihood tree constructed using all samples (without pruning close relatives) produced somewhat different patterns for the Korean and Washington samples (Supplementary Figure S3). In this tree, the Washington samples stood out together with a group of South Korean samples, all of which were collected from the same nest near Andong, North Gyeongsang Province. While this might be taken as evidence that the Washington samples originated from this region, several factors speak against the likelihood of this possibility. First, Andong is not located close to any major international transport or export hubs, which makes it unlikely that hornets could have traveled directly from there to the US. Andong does export agricultural products, including rice and apples, but these goods are primarily shipped to non-US markets26,27. Second, the branch length between the Washington and Andong samples was much longer than that observed for example between the Nanaimo and Japanese samples. Third, the close proximity between the Washington and Andong samples on the all-samples tree was not found on the tree using only distantly related samples (Fig. 3). This indicates that the proximity of these samples was driven by some other factor, such as the fact that the all-sample tree contained a larger number of samples from these two locations than from any other single locality. Finally, while the the Washington samples shared a single mitochondrial haplotype (’Vman-6’) with the samples from Andong, this haplotype was also shared with samples from two other South Korean localities, including Yeongwul-gun, over 70 km north of Andong (Supplementary Table S2).
Compared to the Washington samples, whose exact region of origin proved challenging to discern based on our current data, the two samples found in Nanaimo exhibited high levels of genetic similarity to those sequenced from Japan. We therefore consider it highly likely that these samples originated somewhere in Japan, and as such we endeavoured to uncover the specific locality within Japan from which these samples originated. The positioning of the Nanaimo samples on the SNP-based maximum-likelihood tree (both the the full and relatedness-pruned versions) placed them as outgroups to the mainland Japanese samples, excluding samples from the islands of Sado and Tsushima (Fig. 3, Supplementary Figure S3). However, the ML tree based on mitotypes (which showed good resolution among Japanese samples) clustered the Nanaimo mitotype (Vman-21) together with mitotypes Vman-18 (Ebino), Vman-19 (Yatsushiro), Vman-20 (Kirishima) and Vman-22 (Yakushima Island), all of which are in or near southern Kyūshū, the most southerly of Japan’s four ’mainland’ islands.
Evidence of inbreeding within native and introduced populations
Introduced populations are (at least initially) small and isolated, and as a result are expected to experience significant inbreeding28,29. The resultant inbreeding depression could provide a significant barrier to the establishment of invasive populations. One factor that may counteract this inbreeding depression in social insects is that a single founding female may store the sperm of multiple males in her spermatheca, although this is unlikely to be a major factor in Vespa mandarinia due to the very low rate of polyandry in this species30. Potentially more relevant, historical inbreeding in the source population may mean that that population has had time to adapt to the loss of genetic diversity, for example by purging deleterious recessive alleles31,32, which would reduce the severity of inbreeding depression experienced by invasive offshoots of that source population. We therefore sought to quantify the degree of inbreeding experienced by native and introduced NGH populations.
One way of assessing inbreeding is via the degree of relatedness between individuals within a population. Doing so using a diploid relatedness estimator33, we found that mainland Chinese and Korean populations exhibited a strongly bimodal distribution of relatedness values, with most individuals wholly unrelated save for a subset of closely related individuals (first-order relations collected from the same nests), as expected for an outbred population (Fig. 4). Relatedness among the Japanese samples (which did not include any within-nest replication) did not exhibit this bimodal distribution, but overall relatedness levels were higher than expected for an outbred population spanning a wide geographic area, with a mean and standard error of 0.395±0.01.
Two populations, those from Taiwan and Washington, stood out as exhibiting very high (first-order or greater) levels of relatedness between the majority of individuals, indicative of widespread inbreeding (US: 0.627±0.01; Taiwan: 0.889±0.005; Fig. 4). The extremely high relatedness values observed among the Taiwanese samples is especially surprising, given that this is a native region within which samples were collected from multiple different nests (reflected in the presence of five different mitotypes among these samples; Supplementary Table S2). These patterns remained the same when using a different relatedness estimator (Supplementary Figure S4).
Although these high levels of relatedness may indicate inbreeding, they can also arise from other factors, such as sampling structure. A more robust method of assessing the degrees of recent inbreeding within populations is by measuring the proportion of the genome, fROH, occupied by long runs of homozygosity (ROHs). fROH can be taken as an estimate of the proportion of the genome that is autozygous, i.e. identical due to inheritance from a common ancestor, and is therefore an accurate measure of inbreeding. Because ROHs will tend to be broken up over time, longer ROHs are indicative of more recent inbreeding. We measured fROH across the 6.01M SNPs among our sampled individuals at three different minimum ROH lengths (0.5, 1, and 2Mb).
We found that, at the bottom end of ROH length (0.5 Mb), all populations exhibited fROH values greater than zero but with substantial variation within and between populations (Fig. 5). The Washington population exhibited the highest mean fROH0.5Mb, but a small number of individuals from other populations possessed similarly elevated values: two from Yunnan and one from Japan. Two more samples, from South Korea, exhibited extremely elevated fROH0.5Mb, nearly double that of the next highest sample.
At the upper end of ROH length (2 Mb), which indicates very recent inbreeding, most individuals exhibited an fROH of zero, indicating a complete lack of runs of homozygosity of this length (Fig. 5). Two populations were exceptional in this respect: all samples from Fujian and a subset of nine samples from Washington exhibited fROH2Mb greater than zero. fROH2Mb values among the Fujian samples were uniform, but the Washington samples fell into three groups: those with zero (fROH = 0; n = 16), intermediate (0.02 > fROH > 0; n = 8) and high (fROH > 0.02, n = 1) values of fROH2Mb. Of the ten females collected from the single nest in 2020, over half (5 of 9) exhibited non-zero values of fROH2Mb, including the single individual with fROH2Mb > 0.02. Values were lower for the three nests eradicated in 2021, with 1/3, 2/6, and 1/6 of females, respectively, exhibiting non-zero fROH2Mb. While these sample sizes are too low to be subjected to statistical analysis, our data may reflect a reduction in the proportion of highly inbred individuals on nests found in Washington in 2021 compared to 2020.
Intriguingly, a single sample from South Korea exhibited extremely elevated fROH2Mb, again double that of the next-highest individual, indicative of extreme inbreeding. This sample was collected from Uijeongbu, Gyeonggi Province, which is located within a natural defile that might conceivably act as a reproductive barrier to hornets within this area and could, therefore, explain this sample’s unexpectedly high level of inbreeding.
Patterns of fROH values among populations were qualitatively similar when using only SNPs within each population (as opposed to a combined set of SNPs from all populations together), although absolute values were, as expected, much lower in this case (Supplementary Figure S5).

Selected photos documenting NGH collection and eradication efforts between 2019 and 2021. Dorsal views of (a) worker collected in Nanaimo, BC, September 2019; (b) male collected in Blaine, WA, July 2020; (c) worker collected in Birch Bay, WA, July 2020; (d) worker collected in Yunnan, China. (e) WSDA agents with samples collected during the removal of a nest in Blaine, WA, September 2021, and (f) NGH workers at the entrance of the same nest prior to removal. (g) Hornet hunter removing an NGH nest near a residence, South Korea.

(a) Distribution of V. mandarinia observations across the species’ native range (blue), including samples sequenced as part of this project (orange). Observation data are taken from GBIF2. A single GBIF observation from Madhya Pradesh, India, merited closer observation due to its unusual location and was subsequently excluded as it was deemed a misidentification based on the record image; (b) The two localities in the Pacific Northwest in which V. mandarinia was discovered in 2019. (c) Specific locations in Blaine, WA from which samples were collected for sequencing in 2020–2021.

Midpoint-rooted maximum-likelihood tree using 29 unrelated V. mandarinia samples and 1.03M LD-pruned SNPs, calculated using the GTR+I+G4 model of nucleotide substitution. Nodes with ultrafast bootstrap support > 99% are denoted with asterisks.

Heatmap of PolyRelatedness relatedness coefficients between pairs of female V. mandarinia samples within each population calculated using a diploid relatedness estimator33. Samples from Nanaimo, Canada are not shown, as this population included only two samples.

Proportion of genome occupied by long runs of homozygosity (ROHs) among V. mandarinia samples from different populations at three different minimum lengths of ROH. Elevated values for longer minimum ROH values indicate more recent inbreeding. Note that the scale of the y-axis varies across the three plots.
Discussion
Invasive eusocial hornets represent a serious threat to agriculturally-important honey bees worldwide, as well as to native insects that play key ecological roles such as pollination and pest control. Effective management of such invasions requires that we predict their occurrences and likelihood of establishment. Here we use two short-lived introduced northern giant hornet populations in the American Pacific Northwest as exemplars for such invasions. As well as assessing the geographic origin of these populations, we assess the factors that may contribute to these populations’ ultimate eradication. We find that, while the more successful of the two introduced populations exhibited strong genomic signals of inbreeding, such signals are also present in some long-standing native populations and such inbreeding therefore likely does not present an insurmountable barrier to establishment for this species. Our results suggest that extrinsic factors, rather than inbreeding depression alone, likely contributed to the apparent failure of these introduced NGH populations to become fully established.
One factor that could feasibly have contributed to invasion failure in this case is competition with other local species, but this appears unlikely. Within its native range, NGH is an apex predator with virtually no natural enemies, and preys upon congeners and dominates use of shared resources (e.g. sap flows)3. Two other extrinsic factors, habitat suitability and prey availability, are also unlikely to have contributed to this failure to establish. Species distribution modelling indicates that the Pacific Northwest of North America represents a climatically suitable habitat for NGH22,24, which is reinforced by the initial expansion of the Washington population from one nest in 2020 to three nests in 2021, a time period across which mean temperatures and precipitation in Washington remained relatively stable34. Metabarcoding of larval feces from three of the NGH nests collected in Washington indicates that workers from these nests were able to successfully exploit prey species in the introduced range35. We also note that mate limitation may be a factor in the lack of detections over the past two seasons; in at least one native population, fewer than half of NGH females will accept a mate3, a characteristic whose impact may be exaggerated in a small population. Nonetheless, while we cannot conclusively rule out all extrinsic factors, evidence against a role for climate, competition or lack of prey makes it likely that concerted eradication efforts by local agencies and the public played a critical role in the ongoing extirpation of these introduced populations. Our results thus emphasize the importance of rapid response times and continued vigilance to control future such introductions.
Previous analyses of mitochondrial data revealed two separate North American origins for the Nanaimo and Washington samples, with these populations estimated as having originated from Japan and South Korea respectively14,36. In confirmation of this, we found that whole-genome data from the Nanaimo and Japanese samples that we sequenced exhibited strong similarity to one another while remaining genetically distinct from samples collected elsewhere in NGH’s native range. Patterns of relatedness between our samples from Washington and South Korea were more complex. Although a SNP-based maximum-likelihood tree of distantly-related individuals did place the Washington samples among those from South Korea, confidence estimates for the branching among South Korean samples was generally low (Fig. 3), and PCA placed the Washington and South Korean samples into separate clusters (Supplementary Figure S1). The single mitotype present in the Washington population was shared with a number of samples from Pungcheon-Myeon, South Korea, but this mitotype was also found in a sample from Yeongwul-gun, over 70 km distant from Pungcheon-Myeon. Although we sampled hornets from a wide variety of localities across South Korea (Fig. 2a), the Korean population exhibited a high degree of genetic diversity (Figs. 3, 4), and it is therefore possible that the Washington population originated elsewhere in South Korea. We were also unable to acquire samples for comparison from other, geographically close but harder-to-access regions such as North Korea, far north-east China, and Primorsky Krai, Russia. As such, it is likely that our dataset only captures a fraction of the total genomic variation present across NGH populations worldwide.
A striking result of both PCA and ML analysis was the relatively extreme distance of the mainland Chinese populations from each other and from all other samples. Notably, these samples were taken from populations that at one point were considered Vespa magnifica37, a designation which at present is synonymous with V. mandarinia38 but which, based on the genetic distances observed in our data, might qualify for reconsideration as a genuinely separate species or subspecies. While we sampled only a single nest each from Yunnan and Fujian, the large genetic distances of these nests between one another and with other native samples suggest that a great deal of genetic variation likely exists across the Chinese mainland. Much additional sequencing, especially across northern and central China, will be required if this variation is to be resolved.
Patterns of relatedness and of fROH (the proportion of the genome occupied by runs of homozygosity) indicate that most native populations have experienced a degree of inbreeding, with all individuals exhibiting fROH1000kb greater than 0 (Fig. 5). Elevated fROH typically indicates historical inbreeding, especially in conjunction with the unusually high relatedness coefficients exhibited among individuals from the Taiwanese population. Inbreeding depression represents a significant barrier to the establishment of invasive populations39, but our observation of signatures of historical inbreeding in native NGH populations suggests that this species has already been exposed to a nontrivial degree of historical inbreeding, which could contribute to increased resilience against inbreeding depression (for example via the purging of deleterious recessive alleles40). Nonetheless, only a small number of individuals exhibited nonzero values for the longest value of fROH that we measured (Fig. 5), and almost all of these individuals were from the introduced Washington population. This does therefore indicate that this population experienced a greater degree of inbreeding than is typical in any of the native populations of this species, leaving open the possibility that inbreeding depression played a role in the eventual decline of this population. Notably, the NGH nests collected in Washington were small and appeared to be over-producing males, both of which could be indicators of inbreeding depression19.
Our data indicate that the Washington NGH population was likely founded by a single introduced queen: only a single mitotype was present in this population, and the high level of relatedness within this population compared to the South Korean population that represents its presumed origin is consistent with the possibility of founding by one singly-mated queen. Propagule pressure is usually considered a key factor in determining invasion success41,42, but our results show that for invasive hornets such as NGH a single introduction may be sufficient to begin the invasion process. This reinforces findings from the highly successful invasive V. velutina population in Europe, which likewise began with a single foundress15. The capacity of a single hornet foundress to seed an entire invasive population, even in the face of significant inbreeding, means that intercepting such foundresses in transit should be a top priority in preventing future hornet incursions in the US.
NGH is just one of several hornets that have exhibited invasive potential. At least eight Vespa species have been detected outside of their native ranges, of which five have been documented as establishing long-term non-native populations4. Of these, V. velutina, in addition to being the only other species for which genetic data are available, has been by far the most successful, with invasive populations established in western Europe15, Japan43 and South Korea44. Of particular interest as a comparison to NGH, V. velutina’s invasive range has spread more slowly in South Korea than in France4,45,46, a disparity that may be attributable to competition47: the only native hornet species in France is V. crabro, whereas in South Korea V. velutina must compete with five native Vespa species, four of which (including NGH) outcompete V. velutina in dominance interactions48. If competition with native hornets is a major factor limiting invasive success among hornets, then NGH, which is the largest and most competitively dominant of all hornet species, may be uniquely advantaged as an invader.
Although the two NGH populations introduced to the Pacific North-West in 2019 appear close to being extirpated, the threat of invasive hornets to ecosystems in North America and elsewhere remains high. In August 2023, the United States Department of Agriculture confirmed the identification of a yellow-legged hornet, Vespa velutina, found in Georgia, USA, and by the end of 2023 five nests had been discovered and eradicated. Preventing the establishment of this and other populations of invasive hornets requires a robust understanding of the origins of such populations and the factors that influence their probability of invasive success. The case of NGH in the Pacific Northwest should serve as an exemplar for the successful control of a introduced hornet population. Moreover, as the first whole-genome population genetic dataset available for any hornet species, our data will serve as a foundation for genetic analysis of future hornet invasions.
Methods
Sample collection and sequencing
NGH samples were collected from native and introduced populations from 2019-2022, except for seven samples collected in Japan in earlier years (Supplementary Table S3). For the North American introduced populations, all available samples were sequenced. Because previous analyses of mitochondrial data indicated two separate North American origins, from Japan and South Korea, respectively14, we collected samples from a wide range of localities in those two regions with the hope of identifying the point of origin of the introduced samples more precisely. For the remaining sampled native regions (Fujian, Yunnan, and Taiwan), samples were collected from a single locality. A summary of samples used in this paper is given in Table 1 and more complete details are provided in Supplementary Table S3.
Thoracic muscle tissue was extracted from collected samples using the MasterPure Complete DNA and RNA Purification Kit (Epicentre, Madison, WI). Library preparation was performed by Novogene Co. followed by sequencing on an Illumina NovaSeq 6000 platform with 30–50M 150-bp paired-end reads/sample.
Read alignment and variant calling
We trimmed sequenced reads of low-quality sequences using Trimmomatic v0.3949. We aligned trimmed reads to the GCF_014083535.2 NGH RefSeq assembly using NextGenMap v0.5.2 with default parameters. Duplicated reads were marked and removed using GATK v4.2.2.0. GATK was then used to filter SNPs using the following filters: QD < 2.0; QUAL < 30.0; SOR > 3.0; FS > 60.0; MQ < 40.0; MQRankSum \(< -12.5\); ReadPosRankSum \(< -8.0\).
Relatedness estimation
We estimated the relatedness between samples using PolyRelatedness v1.1150. To prepare the dataset, we filtered the raw variants using VCFtools, retaining genotype calls with a minimum quality score of 20 and removing loci with 10 percent missing data. We removed loci with a read depth of two or fewer to obtain loci suitable for parentage and relatedness analysis. We then used PLINK v2.051 to filter one locus of each pair in a sliding 200-kb window using a threshold r2>0.1, resulting in a final set of 111,215 unlinked loci. To reduce the number of loci to a manageable amount for PolyRelatedness, we sampled every 90th locus and included those in the dataset for running the program. Finally, we ran PolyRelatedness on only female samples using a diploid relatedness estimator33.
Detection of runs of homozygosity
In line with published recommendations52, we performed ROH analyses using SNPs un-filtered for LD. Male hornets are hemizygous and were therefore excluded from ROH analyses. All ROH estimates were generated in PLINK v2.051 using the—homozyg argument with default settings except for the following:—homozyg-snp 50;—homozyg-density 60;—homozyg-gap 500; and—homozyg-window-het 1. Each ROH analysis was performed once each for minimum ROH lengths of 500 kb, 1000 kb, and 2000 kb.
For a given set of SNPs and minimum length of ROH, we first generated synthetic data for a hypothetical individual homozygous for every one of those SNPs, and then defined (ROHmax) as the total length of ROHs that would be detected in this fully-homozygous individual. This value represented the theoretical upper bound for ROHs in any individual measured at this set of SNPs. We then defined ROHtrue as the length of ROHs identified using PLINK in each real sample for the same set of SNPs, and defined fROH for that sample and minimum ROH length as ROHtrue/ROHmax. In this way, for example, fROH500kb for a given sample represents the length of that individual’s genome found within ROHs of 500kb or larger, relative to the maximum length of genome that could theoretically be found within such ROHs.
Estimating fROH relative to a theoretical maximum ROH for a given set of SNPs corrected for the fact that ROHs can only be detected within a certain genomic region if the density of SNPs in that region is sufficiently high. However, the choice of SNPs used may introduce biases, for example when a site is fixed for two different alleles across two populations, such that it is polymorphic when considered across the two populations but is monomorphic (and therefore always homozygous) when looking within a single population. To account for this possibility, we generated fROH values in each population twice: once using the full set of SNPs discovered across all populations, and again for each population using only SNPs that were polymorphic within that population.
Principal component analyses and maximum-likelihood trees
To determine the genetic structure of populations in each species across their range, we used principal component analyses (PCAs) and maximum-likelihood (ML) trees. Prior to these analyses, SNPs were filtered to exclude sites in strong linkage disequilibrium (LD) with one another. We used PLINK v2.051 to remove SNPs with pairwise r2>0.8 within sliding windows of 50 SNPs, with a 5-SNP increment between windows, a standard parameterization53.
We performed principal component analysis using the same linkage-pruned datasets in R v4.1.254 using the snpgdsPCA() function, part of the package SNPRelate v1.26.055. Because the initial PCA showed the two mainland Chinese populations to be highly distinct from all other populations, such that it was not possible to resolve the clustering of samples among and within the other populations, we also generated additional PCAs without the mainland Chinese samples.
Some of the samples in our dataset were closely related to one another, for example because they were sampled from the same nest. Our initial testing found that including many close relatives with very short branch lengths resulted in ML trees that were very crowded and inflated the branch lengths to clusters of close relatives. We, therefore, pruned our data of closely related individuals before the construction of ML trees using NAToRA56 together with the relatedness values generated using PolyRelatedness. Using a relatedness cutoff of 0.33, the harmonic mean of 0.5 and 0.2 (relatedness values denoting first- and second-order relatives, respectively), we pruned our dataset down to 29 distantly-related individuals.
We constructed ML trees using relatedness- and LD-filtered SNP data in R v4.1.254. Data were read into R using vcfR v1.14.057 and converted to ML trees using ape v5.758 and phangorn v2.11.1. To speed computation, trees were generated using a subset of 10,000 SNPs. Model suitability was assessed using the modelTest() function and a GTR+G(4)+I nucleotide substitution model was selected as optimal based on BIC values. Bootstrap values were obtained using 1000 ultrafast bootstrap replicates59, as implemented by phangorn. Tree files were converted to Newick format and plotted using FigTree v.1.4.460. For readability, bootstrap values in the figures were binarized, with nodes with bootstrap support > 99% denoted with an asterisk (*).
Mitochondrial super-barcoding
To further examine the origins and genetic diversity of introduced NGH to Washington state, we applied a super barcode approach61,62 utilizing mitogenomic data from the whole genome shotgun data to quantify the number of foundresses involved with the initial introduction. To do this, reads from the mitogenome were extracted from the whole genome data by comparison to the V. mandarinia reference mitogenome NC_050197.1. The reads were then assembled into consensus sequences for each individual using BCFtools 1.863, with missing alleles set to N. To ensure high- quality SNV calls were used in haplotype identification, regions in the consensus sequences with less than 100x mean read depth were removed. The alignment was further trimmed by removing first individuals with more than 0.4% Ns (indicative of low individual coverage and/or poor-quality template DNA) and then all loci across all remaining individuals that contained any Ns or ambiguous bases. Proceeding trimming and after each trimming step, alignments were generated using MAFFT 7.45064,65. Annotated NGH mitogenome reference OQ836204.1 was included through the above steps to assess the genic location of SNVs and ensure that the data set generated in this study was not frameshifted in relation to the reference due to the above trimming steps. The trimmed sequences were also back- checked by mapping the trimmed sequences to the untrimmed reference. The trimmed and checked alignment was then used to generate a Maximum Likelihood (ML) phylogenetic tree using IQ-TREE66 with default settings, model selection using ModelFinder67 and clade support assessed using ultrafast bootstrapping59 as well as the nonparametric SH-aLRT68,69,70. Based on these results, a Bayesian inference (BI) tree was resolved using our samples from Yunnan as an outgroup, given their genetic distance from the remaining samples and their closest match to V. magnifica in BLASTn. The program MrBayes 3.2.671 was employed to conduct Bayesian inference and was run with a general time reversible + invariable sites + gamma rate variation among sites substitution and rate variation model, four gamma categories, six heated chains, a chain length of 1,100,000, a subsampling frequency of 200, a burn-in of 100,000, and an unconstrained branch length prior. Finally, a TCS statistical parsimony72 network was generated from the alignment using PopART 1.773. Haplotype calls and sequence characterizations were implemented in DNAsp 6.12.0374.
Data availibility
Raw sequencing data are available at the Sequence Read Archive (SRA) under BioProject PRJNA1069611.
References
Archer, M. E. Taxonomy, distribution and nesting biology of the Vespa mandarinia group (Hym., Vespidae). Entomol. Month. Mag. 131, 47–53 (1995).
GBIF.org. Occurrence Download, https://doi.org/10.15468/DL.HJXUQW (2023).
Matsuura, M. & Yamane, S. Biology of the Vespine Wasps (Springer, 1990).
Otis, G. W., Taylor, B. A. & Mattila, H. R. Invasion potential of hornets (Hymenoptera: Vespidae: Vespa spp.). Front. Insect Sci. 3, 1145158 (2023).
Matsuura, M. Ecological study on Vespine Wasps (Hymenoptera:Vespidae) attacking honeybee colonies: I. Seasonal changes in the frequency of visits to Apiaries by Vespine Wasps and damage inflicted, especially in the absence of artificial protection. Appl. Entomol. Zool. 23, 428–440. https://doi.org/10.1303/aez.23.428 (1988).
Matsuura, M. & Sakagami, S. A Bionomic Sketch of the Giant Hornet, Vespa mandarinia, a Serious Pest for Japanese Apiculture. J. Fac. Sci. Hokkaido University Series VI. Zoology. 19, 25-162 (1973).
Tan, K. et al. Honey bee inhibitory signaling is tuned to threat severity and can act as a colony alarm signal. PLoS Biol. 14, e1002423. https://doi.org/10.1371/journal.pbio.1002423 (2016).
Fujiwara, A., Sasaki, M. & Washitani, I. First report on the emergency dance of Apis cerana japonica, which induces odorous plant material collection in response to Vespa mandarinia japonica scouting. Entomol. Sci. 21, 93–96. https://doi.org/10.1111/ens.12285 (2018).
Ono, M., Igarashi, T., Ohno, E. & Sasaki, M. Unusual thermal defence by a honeybee against mass attack by hornets. Nature 377, 334–336. https://doi.org/10.1038/377334a0 (1995).
McClenaghan, B. et al. Behavioral responses of honey bees, Apis cerana and Apis mellifera, to Vespa mandarinia marking and alarm pheromones. J. Apicult. Res. 58, 141–148. https://doi.org/10.1080/00218839.2018.1494917 (2019).
Mattu, V. & Sharma, I. Seasonal variation of Vespa auraria S. and Vespa mandarinia S. (Hymenoptera: Vespidae) attacking Apis mellifera L. colonies in district Kangra of Western Himalayas, Himachal Pradesh (India). J. Entomol. Zool. Stud. 5, 1862-1864 (2017).
Bertelsmeier, C. Globalization and the anthropogenic spread of invasive social insects. Curr. Opin. Insect Sci. 46, 16–23. https://doi.org/10.1016/j.cois.2021.01.006 (2021).
Goodisman, M. A. D. The curse of invasive social insects. Insectes Soc. 70, 283–284. https://doi.org/10.1007/s00040-023-00932-5 (2023).
Wilson, T. M., Takahashi, J., Spichiger, S.-E., Kim, I. & van Westendorp, P. First reports of Vespa mandarinia (Hymenoptera: Vespidae) in North America represent two separate maternal lineages in Washington State, United States, and British Columbia, Canada. Ann. Entomol. Soc. Am. 113, 468–472. https://doi.org/10.1093/aesa/saaa024 (2020).
Arca, M. et al. Reconstructing the invasion and the demographic history of the yellow-legged hornet, Vespa velutina, Europe. Biol. Invasions 17, 2357–2371. https://doi.org/10.1007/s10530-015-0880-9 (2015).
Dlugosch, K. M., Anderson, S. R., Braasch, J., Cang, F. A. & Gillette, H. D. The devil is in the details: Genetic variation in introduced populations and its contributions to invasion. Invasion genetics: The baker and stebbins legacy 232–251 (2016).
Taylor, C. M. & Hastings, A. Allee effects in biological invasions. Ecol. Lett. 8, 895–908 (2005).
Berube, C. Giant alien insect invasion averted. Am. Bee J. 160, 209–214 (2020).
Looney, C. et al. Detection and description of four Vespa mandarinia (hymenoptera: Vespidae) nests in western North America. J. Hymenopt. Res. 96, 1–20 (2023).
Stephens, P. A., Sutherland, W. J. & Freckleton, R. P. What is the Allee effect?. Oikos 87, 185–190. https://doi.org/10.2307/3547011 (1999).
Kramer, A. M., Dennis, B., Liebhold, A. M. & Drake, J. M. The evidence for Allee effects. Popul. Ecol. 51, 341–354. https://doi.org/10.1007/s10144-009-0152-6 (2009).
Zhu, G., Gutierrez Illan, J., Looney, C. & Crowder, D. W. Assessing the ecological niche and invasion potential of the Asian giant hornet. Proc. Natl. Acad. Sci. 117, 24646–24648. https://doi.org/10.1073/pnas.2011441117 (2020).
Moo-Llanes, D. A. Inferring distributional shifts of asian giant hornet Vespa mandarinia Smith in climate change scenarios. Neotrop. Entomol. 50, 673–676. https://doi.org/10.1007/s13744-020-00840-4 (2021).
Nuñez-Penichet, C. et al. Geographic potential of the world’s largest hornet, Vespa mandarinia Smith (Hymenoptera: Vespidae), worldwide and particularly in North America. PeerJ 9, e10690. https://doi.org/10.7717/peerj.10690 (2021).
Song, K., Li, L. & Zhang, G. Coverage recommendation for genotyping analysis of highly heterologous species using next-generation sequencing technology. Sci. Rep. 6, 35736 (2016).
Korea JoongAng Daily. Korean rice gains ground overseas. https://koreajoongangdaily.joins.com/2009/08/17/features/Korean-rice-gains-ground-overseas/2908882.html (2009).
ExportHub. Andong Trading Corporation. https://www.exporthub.com/andong-trading-corporation/ (2024).
Kirkpatrick, M. & Jarne, P. The effects of a bottleneck on inbreeding depression and the genetic load. Am. Nat. 155, 154–167. https://doi.org/10.1086/303312 (2000).
Wright, L. I., Tregenza, T. & Hosken, D. J. Inbreeding, inbreeding depression and extinction. Conserv. Genet. 9, 833–843. https://doi.org/10.1007/s10592-007-9405-0 (2008).
Takahashi, J.-I., Akimoto, S., Martin, S. J., Tamukae, M. & Hasegawa, E. Mating structure and male production in the giant hornet Vespa mandarinia (Hymenoptera: Vespidae). Appl. Entomol. Zool. 39, 343–349. https://doi.org/10.1303/aez.2004.343 (2004).
Lee, C. E. Evolutionary genetics of invasive species. Trends Ecol. Evol. 17, 386–391. https://doi.org/10.1016/S0169-5347(02)02554-5 (2002).
Hedrick, P. W. & Garcia-Dorado, A. Understanding inbreeding depression, purging, and genetic rescue. Trends Ecol. Evol. 31, 940–952. https://doi.org/10.1016/j.tree.2016.09.005 (2016).
Anderson, A. D. & Weir, B. S. A maximum-likelihood method for the estimation of pairwise relatedness in structured populations. Genetics 176, 421–440 (2007).
National Oceanic and Atmospheric Administration National Centers for Environmental information Climate at a Glance: Statewide Time Series. Available at https://www.ncei.noaa.gov/access/monitoring/climate-at-a-glance/statewide/time-series (2024/04/22).
Wilson, T. et al. Insights into the prey of Vespa mandarinia (Hymenoptera: Vespidae) in Washington state, obtained from metabarcoding of larval feces. Front. Insect Sci. 3, 1134781 (2023).
Freeman, A. & Xia, X. Phylogeographic reconstruction to trace the source population of Asian giant hornet caught in Nanaimo in Canada and Blaine in the USA. Life 14, 283 (2024).
Archer, M. E. Vespine wasps of the world: Behaviour, ecology and taxonomy of the Vespinae. Manchester: Siri Scientific Press. (2012).
Smith-Pardo, A. H., Carpenter, J. M. & Kimsey, L. The diversity of hornets in the genus Vespa (Hymenoptera: Vespidae; Vespinae), their importance and interceptions in the United States. Insect Syst. Divers. 4, 2. https://doi.org/10.1093/isd/ixaa006 (2020).
Fauvergue, X., Vercken, E., Malausa, T. & Hufbauer, R. A. The biology of small, introduced populations, with special reference to biological control. Evolut. Appl. 5, 424–443. https://doi.org/10.1111/j.1752-4571.2012.00272.x (2012).
Crnokrak, P. & Barrett, S. C. H. Perspective: Purging the genetic load: A review of the experimental evidence. Evolution 56, 2347–2358. https://doi.org/10.1111/j.0014-3820.2002.tb00160.x (2002).
Lockwood, J. L., Cassey, P. & Blackburn, T. M. The more you introduce the more you get: The role of colonization pressure and propagule pressure in invasion ecology. Divers. Distrib. 15, 904–910. https://doi.org/10.1111/j.1472-4642.2009.00594.x (2009).
Simberloff, D. The role of propagule pressure in biological invasions. Annu. Rev. Ecol. Evol. Syst. 40, 81–102. https://doi.org/10.1146/annurev.ecolsys.110308.120304 (2009).
Kishi, S. & Goka, K. Review of the invasive yellow-legged hornet, Vespa velutina nigrithorax (hymenoptera: Vespidae), in Japan and its possible chemical control. Appl. Entomol. Zool. 52, 361–368 (2017).
Choi, M. B., Martin, S. J. & Lee, J. W. Distribution, spread, and impact of the invasive hornet Vespa velutina in South Korea. J. Asia-Pac. Entomol. 15, 473–477 (2012).
Jung C. et al. Spatial expansion of an invasive hornet, Vespa velutina nigrithorax buysson (Hymenoptera: Vespidae) in Korea. Korean J. Apic. 27, 87-93 (2012).
Robinet, C., Suppo, C. & Darrouzet, E. Rapid spread of the invasive yellow-legged hornet in France: The role of human-mediated dispersal and the effects of control measures. J. Appl. Ecol. 54, 205–215 (2017).
Villemant, C. et al. Predicting the invasion risk by the alien bee-hawking yellow-legged hornet Vespa velutina nigrithorax across Europe and other continents with niche models. Biol. Cons. 144, 2142–2150 (2011).
Kwon, O. & Choi, M. B. Interspecific hierarchies from aggressiveness and body size among the invasive alien hornet, Vespa velutina nigrithorax, and five native hornets in South Korea. PLoS ONE 15, e0226934 (2020).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Huang, K., Ritland, K., Guo, S., Shattuck, M. & Li, B. A pairwise relatedness estimator for polyploids. Mol. Ecol. Resour. 14, 734–744. https://doi.org/10.1111/1755-0998.12217 (2014).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaSciencehttps://doi.org/10.1186/s13742-015-0047-8 (2015).
Meyermans, R., Gorssen, W., Buys, N. & Janssens, S. How to study runs of homozygosity using plink? A guide for analyzing medium density snp data in livestock and pet species. BMC Genom. 21, 1–14 (2020).
Novembre, J. et al. Genes mirror geography within Europe. Nature 456, 98–101. https://doi.org/10.1038/nature07331 (2008).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria (2022).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328. https://doi.org/10.1093/bioinformatics/bts606 (2012).
Leal, T. P. et al. NAToRA, a relatedness-pruning method to minimize the loss of dataset size in genetic and omics analyses. Comput. Struct. Biotechnol. J. 20, 1821–1828. https://doi.org/10.1016/j.csbj.2022.04.009 (2022).
Knaus, B. J. & Grünwald, N. J. VCFR: A package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53 (2017).
Paradis, E. & Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. https://doi.org/10.1093/bioinformatics/bty633 (2019).
Minh, B. Q., Nguyen, M. A. T. & Von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195 (2013).
Rambaut, A. Figtree tree figure drawing tool. http://tree.bio.ed.ac.uk/software/figtree/ (2009).
Li, X. et al. Plant DNA barcoding: From gene to genome. Biol. Rev. 90, 157–166. https://doi.org/10.1111/brv.12104 (2015).
Dziedzic, E. et al. Creating, curating and evaluating a mitogenomic reference database to improve regional species identification using environmental DNA. Mol. Ecol. Resour. 23, 1880–1904. https://doi.org/10.1111/1755-0998.13855 (2023).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008. https://doi.org/10.1093/gigascience/giab008 (2021).
Katoh, K., Misawa, K., Kuma, K.-I. & Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. https://doi.org/10.1093/nar/gkf436 (2002).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. https://doi.org/10.1093/molbev/msu300 (2015).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. https://doi.org/10.1038/nmeth.4285 (2017).
Kishino, H. & Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J. Mol. Evol. 29, 170–179. https://doi.org/10.1007/BF02100115 (1989).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. https://doi.org/10.1093/sysbio/syq010 (2010).
Shimodaira, H. & Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16, 1114 (1999).
Huelsenbeck, J. P. & Ronquist, F. Mrbayes: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Clement, M., Posada, D. & Crandall, K. TCS: A computer program to estimate gene genealogies. Mol. Ecol 9, 1657–1659 (1999).
Leigh, J. W. & Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. https://doi.org/10.1093/molbev/msx248 (2017).
Acknowledgements
This work was made possible, in part, by Cooperative Agreements from the United States Department of Agriculture’s Animal and Plant Health Inspection Service (APHIS). It may not necessarily express APHIS’ views. B.A.T is additionally supported by a Human Frontier Science Program fellowship (ID 885512). We would like to thank Dr. Moon-Bo Choi, Cheol-Ho Ma, Jeong-An Kim, Yeon-Guk Choi, Mr. Ahn, Dr. Yue-Wen Chen, and Dr. Chung-Yu Ko for contributing specimens used in this study.
Author information
Authors and Affiliations
Contributions
B.A.T., L.R.T. and M.S. performed the analyses and generated figures, with input from B.A.H. B.A.T., T.M.W., C.L., A.H.S.P., T.M.G., J.T., and B.A.H. coordinated sample collection and sequencing. C.L. collected samples from South Korea. B.A.T. wrote the main manuscript text with input from all authors. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taylor, B.A., Tembrock, L.R., Sankovitz, M. et al. Population genomics of the invasive Northern Giant Hornet Vespa mandarinia in North America and across its native range. Sci Rep 14, 10803 (2024). https://doi.org/10.1038/s41598-024-61534-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-61534-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
