
Abstract
Cyclin-dependent kinases (CDKs) 4 and 6 inhibitors (CDK4/6is) are effective in metastatic breast cancer, but they have been only modestly effective in most other tumor types. Here we show that tumors expressing low CDK6 rely on CDK4 function and are exquisitely sensitive to CDK4/6is. In contrast, tumor cells expressing both CDK4 and CDK6 have increased reliance on CDK6 to ensure cell cycle progression. We discovered that CDK4/6is and CDK4/6 degraders potently bind and inhibit CDK6 selectively in tumors in which CDK6 is highly thermo-unstable and strongly associated with the HSP90–CDC37 complex. In contrast, CDK4/6is and CDK4/6 degraders are ineffective in antagonizing tumor cells expressing thermostable CDK6, due to their weaker binding to CDK6 in these cells. Thus, we uncover a general mechanism of intrinsic resistance to CDK4/6is and CDK4/6i-derived degraders and the need for new inhibitors targeting the CDK4/6i-resistant, thermostable form of CDK6 for application as cancer therapeutics.
Similar content being viewed by others

Main
CDK4/6 regulate cell cycle progression by phosphorylating and inactivating the tumor suppressor retinoblastoma protein (RB) and thus have been targeted by small-molecule inhibitors for cancer therapy1,2. Dissociation of hyper-phosphorylated RB alleviates transcriptional repression of E2F promoters and allows initiation of DNA synthesis- and mitosis-related gene transcription2,3. Recently, CDK4/6is in combination with hormonal therapy showed significant clinical activity in RB-proficient metastatic ER+ breast cancers4,5 and three CDK4/6is, palbociclib, abemaciclib and ribociclib, are now US Food and Drug Administration (FDA)-approved for this indication4,6,7,8. As the activity of CDK4/6 requires a functional RB protein, tumors that do not express functional RB are resistant to these drugs9. However, in many tumor types predominantly expressing wild-type (WT) RB1 (lung adenocarcinomas, melanomas, colon cancers and others) preclinical and clinical studies have shown only modest effectiveness of CDK4/6is10,11,12, suggesting that other mechanisms limit their efficacy in these tumor types. In ER+ breast cancer, CDK6 amplification has been reported to confer acquired resistance to CDK4/6is13. However, CDK4/6is are potent inhibitors of CDK6 in vitro and they show effectiveness in some, mostly liquid, tumor types predominantly expressing CDK6 (ref. 14). In solid tumors a comprehensive analysis of CDK6 expression at baseline and its potential role in tumor response to CDK4/6is has been lacking. Thus, there is a need for better understanding of the role of CDK6 as a potential contributor to tumor resistance to CDK4/6is.
Similarly to most kinases, CDK4 and, to a lesser extent, CDK6 has been previously shown to require the HSP90–CDC37 chaperoning complex for their maturation15,16,17. Closely related kinases may show largely different dependencies on HSP90–CDC37, for example, CDK4 is highly dependent on HSP90, but the closely related CDK2 is almost completely independent18. Further, activating mutations have been found to increase the dependency of kinases19,20, but overall, dependence of a given kinase on HSP90–CDC37 is thought to be uniform across cells and to depend exclusively on its amino-acid sequence. Finally, small-molecule inhibitors have been shown to compete with HSP90–CDC37 for kinase binding18 and there is evidence from primarily in vitro studies for a connection between the strength of the interaction of CDKs with the HSP90–CDC37 complex and their affinity for inhibitors21. However, whether the kinase interaction with the HSP90–CDC37 complex affects tumor response to small-molecule inhibitors remains unknown.
Lately, proteolysis-targeted chimeras (PROTACs), which are hetero-bifunctional small molecules that are aimed at achieving selective degradation of the target proteins have been developed against a number of target kinases, including CDK4/6 (refs. 22,23,24). We developed a potent and selective CDK4/6-directed PROTAC (CDK4/6 degrader) and we used it as tool to elucidate mechanisms of CDK4/6 regulation and response to CDK4/6is, by monitoring target degradation by CDK4/6 degrader as a surrogate for compound binding to target in cells. Use of this approach provided us with evidence of a critical role of the expression state of CDK6 in affecting tumor response to CDK4/6is.
Results
Intrinsic resistance to CDK4/6is is associated with incomplete inhibition of RB–E2F and expression of CDK6
To gain insight into mechanisms of intrinsic resistance or sensitivity to CDK4/6is, we assessed the concentration-dependent effects of palbociclib on the growth of cancer cell lines derived from a variety of RB-proficient tumor types. We observed large variations in cell line response to CDK4/6is, consistent with previous reports11,12,25. In the ‘CDK4/6i-sensitive’ (CDK4/6i-S) group, palbociclib concentrations <500 nM and in many cases as low as 80 nM were sufficient for substantial (>90%) growth inhibition. In contrast, in the ‘CDK4/6i-resistant’ (CDK4/6i-R) group, palbociclib concentrations even as high as 2 μM had only modest effects on cell growth (Fig. 1a and Extended Data Fig. 1a). The CDK4/6i-S group included ER+ breast cancer cell lines, as expected26, as well as cancer cell lines from several other tumor types. However, most RB-proficient cell lines derived from common solid tumor types, including nonsmall cell lung carcinoma (NSCLC), melanoma and colorectal carcinoma showed a CDK4/6i-R phenotype (Fig. 1a and Extended Data Fig. 1a).

a, Bar graph of GI90 values based on cell growth crystal violet assays for the indicated cell lines treated with increasing concentrations of palbociclib (PB) for 10–15 d. CDK4/6i-S cell lines are highlighted in blue and CDK4/6i-R cell lines are in red. GI90, 90% cell growth inhibition. b, The indicated cell lines were treated with 1 µM palbociclib for 24, 48 and 72 h and lysates were immunoblotted with the indicated antibodies. c, The indicated cell lines were treated with 3 µM ribociclib for 24, 48 and 72 h and lysates were immunoblotted with the indicated antibodies. d, The indicated cell lines were treated with 0.3 µM abemaciclib (AB) for 24, 48 and 72 h and lysates were immunoblotted with the indicated antibodies. e, Expression of known cell cycle regulators was detected by immunoblotting with the indicated antibodies. All data represent single experiments.
We next assessed whether the observed differences of tumor cell sensitivity to palbociclib were associated with the extent of suppression of RB–E2F output by the drug. Indeed, suppression of both RB phosphorylation and of downstream E2F target proteins, PLK1, cyclin A and DHFR upon palbociclib treatment (RB–E2F output) were consistently of greater magnitude in CDK4/6i-S compared to CDK4/6i-R tumor cells (Fig. 1b and Extended Data Fig. 1b,c). Other US FDA-approved CDK4/6is, including ribociclib8 and abemaciclib7, showed a similar profile (Fig. 1c,d). When we examined expression of several known CDK4/6 signaling-related proteins, including members of the cyclin D and cyclin E, INK4 and CIP/KIP families, we did not detect any consistent difference in expression between the two groups (Fig. 1e). However, we noticed that most CDK4/6i-S cell lines expressed CDK6 at very low levels, whereas the CDK4/6i-R cell lines in our panel, expressed both CDK4 and CDK6 (Fig. 1e).
Low expression of CDK6 predicts for sensitivity to CDK4/6is in NSCLC
The observation that low CDK6 expression correlated with sensitivity to CDK4/6is, prompted us to assess whether we could identify subsets of CDK4/6i-S cell lines within larger, predominantly RB-proficient, tumor types. We focused on NSCLC, in which previous studies have shown clinical activity of these drugs, but failed to show a significant increase in either progression-free survival (PFS) or overall survival (OS), over current standard of care27,28. In addition to the NSCLC lines that we examined initially (Extended Data Fig. 1a), we assessed five additional NSCLC lines with a high score for CDK4 dependence (H1792, H2087, H2291, HCC827 and H1915, in addition to H358) based on Achilles RNAi DEMETER scores29. For comparison, we examined two additional NSCLC CDK6-dependent lines (PC9 and H1666, in addition to A549, Calu6). We first confirmed dependence of this NSCLC panel on either CDK4 or CDK6. In CDK4-dependent cells, short interfering (si)RNA-mediated knockdown of CDK4, but not CDK6 suppressed RB–E2F output, whereas downregulation of CDK6, but not of CDK4 suppressed pRB–E2F output in CDK6-dependent cells (Fig. 2a). Further, although expression of CDK4 was variable across our panel of cell lines, CDK4-dependent cells expressed low levels of CDK6 compared to CDK4 (Fig. 2b).

a, The indicated NSCLC cell lines were transfected with control siRNA, siCDK4 and siCDK6 for 72 h and lysates were immunoblotted with the indicated antibodies. b, Total cell lysates of the indicated NSCLC cell lines were subjected to immunoblotting with the indicated antibodies. The ratio of CDK4 to CDK6 was calculated by band intensity using ImageJ. The samples are derived from the same experiment and the images were processed in parallel. c, The indicated NSCLC cell lines were treated with increasing concentrations of PB for 24 h and lysates were immunoblotted with the indicated antibodies. d, Cell growth crystal violet assay for the indicated NSCLC cell lines treated with increasing concentrations of PB for 10–15 d and stained with crystal violet. CDK4/6i-S cell lines are in blue and CDK4/6i-R cell lines are in red. e, PFS analysis of NSCLC/RAS-mutant patients that received AB in the JUNIPER trial based on CDK6-low versus CDK6-high tumors (AB, n = 79 patients; n = 43 patients in CDK6-low arm, n = 36 patients in CDK6-high arm; erlotinib, n = 69 patients; n = 37 patients in CDK6-low arm, n = 32 patients in CDK6-high arm). f, OS analysis of patients with NSCLC/RAS mutation that received AB in the JUNIPER trial based on CDK6-low versus CDK6-high tumors (AB, n = 79 patients; n = 43 patients in CDK6-low arm, n = 36 patients in CDK6-high arm; erlotinib, n = 69 patients; n = 37 patients in CDK6-low arm, n = 32 patients in CDK6-high arm). All data represent single experiments. CI, confidence interval; HR, hazard ratio.
We next assessed whether response to CDK4/6is correlates with low expression of CDK6 in NSCLC tumor cells. In all cases, we observed substantially higher sensitivity of CDK6-low cells to CDK4/6is, both in terms of RB–E2F output (Fig. 2c and Extended Data Fig. 2a) and of cell growth (Fig. 2d) compared to CDK4/6i-R cells with high levels of CDK6 (H358 and A549, Calu6 data are included in Fig. 1a,b and Extended Data Fig. 1a, and a list of the NSCLC cell lines used in our study is in Extended Data Fig. 2b).
To assess the clinical relevance of our observations, we analyzed RNA expression data from tumors from the phase III JUNIPER trial (NCT02152631), in which abemaciclib was evaluated in patients with KRAS-mutated, advanced NSCLC27. In a retrospective assessment, tumors with low CDK6 expression correlated with significantly longer PFS (Fig. 2e) and OS (Fig. 2f) compared to tumors expressing high levels of CDK6. These data confirm in a large clinical dataset, a critical role of CDK6 expression in the tumor in determining outcome of patients with cancer treated with CDK4/6is and suggest that CDK6 expression in the tumor might be used a biomarker to stratify patients with NSCLC for CDK4/6i treatment.
Tumors expressing both CDK4 and CDK6 depend selectively on CDK6
We next focused on understanding the basis of resistance of tumor cells expressing both CDK4 and CDK6 to CDK4/6is. Overexpression of a target is a mechanism that can drive resistance to small-molecule inhibitors. We thus assessed whether short hairpin (sh)RNA-mediated knockdown of CDK4 or CDK6 would sensitize palbociclib-unresponsive cells to CDK4/6is. As expected, in cell lines expressing predominantly CDK4 and low levels of CDK6, knockdown of CDK4, but not of CDK6, reduced RB–E2F output (Extended Data Fig. 3a). In CDK4/6i-R tumor cells, inducible CDK6 knockdown suppressed RB–E2F output (Fig. 3a), as well as cell growth (Fig. 3b), which were further suppressed by treatment with palbociclib. Notably, knockdown of CDK4 in these same cells had no effect on either phosporylated RB (pRB) levels or sensitivity to palbociclib (Fig. 3a,b). CDK6 (but not CDK4) knockdown also suppressed RB–E2F output as well as growth in additional CDK4/6i-R cell lines (Fig. 3c). Moreover, ectopic expression of an shRNA-resistant mutant of CDK6 rescued cell growth and RB–E2F output inhibition promoted by CDK6 shRNA-mediated knockdown (Fig. 3d), thus establishing the specific role of CDK6 in driving RB–E2F signaling and cell growth in these cells. Finally, phosphorylation of CDK4-T172, an established marker of CDK4 activation30, was readily detected in the CDK4/6i-S cells, but was substantially lower in the CDK4/6i-R cells (Fig. 1e). All of these findings suggested that despite expression of both CDK4 and CDK6, RB–E2F signaling was driven selectively by CDK6 in CDK4/6i-R tumor cells.

a, A549 and A375 expressing doxycycline (Dox)-inducible shCDK4 or shCDK6 were treated with 0.1 µg ml−1 Dox for 36 h, followed by PB for 24 h. Cell lysates were immunoblotted with the indicated antibodies. b, Cell growth crystal violet assay for A549 and A375 expressing Dox-inducible shCDK4 or shCDK6 in the presence or absence of 0.1 µg ml−1 Dox and increasing concentrations of PB for 10 d. c, The indicated cell lines expressing Dox-inducible shCDK4 or shCDK6 were treated with or without 0.1 µg ml−1 Dox for 10 d followed by crystal violet staining (top). Cell lines were treated with or without 0.1 µg ml−1 Dox for 72 h and cell lysates were subjected to immunoblotting with the indicated antibodies (bottom). d, The indicated cell lines coexpressing Dox-inducible shCDK6 and shRNA-resistant form of V5-CDK6 (shR-CDK6) were treated with or without 0.05 µg ml−1 Dox for 10 d followed by crystal violet staining (top). Cell lines were treated with or without 0.05 µg ml−1 Dox for 72 h and cell lysates were subjected to immunoblotting with the indicated antibodies (bottom). e, Scatter-plots of DEMETER score (DepMap RNAi; DEMETER2 Data v.5) and expression for CDK4 and CDK6 (top). Scatter-plots of CERES (DepMap CRISPR; Public 20Q1) score and MS-based proteomics levels of CDK4 and CDK6 (ref. 35) (bottom). All expression values are in log2(TPM + 1). TPM, transcripts per million. Proteomic levels are shown as normalized log2-transformed ratios to the bridge sample in each tandem mass tag 10-plex as previously62 described. Cell lines harboring COSMIC hotspot mutations to RB1 are annotated in orange. P values were calculated based on linear regression analysis. All data represent single experiments.
To assess the generality of our observation, we analyzed the Avana dependency map (DepMap) dataset to look at the correlation between the expression and protein levels to the dependency profiles of CDK4 and CDK631,32,33,34,35,36. First, similar to the cell lines used in our study, CDK4 expression was relatively uniform across cancer cell lines, whereas CDK6 expression was highly variable. Further, CDK6 expression and protein levels were correlated with dependence on CDK6, however CDK4 protein or mRNA expression levels were not predictive of dependence on CDK4 (Fig. 3e and Extended Data Fig. 3b). The data were similar whether dependency was interrogated using shRNA or single guide (sg)RNA-mediated loss of function31. Thus, the analysis revealed that our observation applies across tumor lines of various cancer types.
Development of MS140, a potent and selective CDK4/6 degrader (PROTAC)
The inactivity of CDK4/6is in cells expressing both CDK4 and CDK6, led us to postulate that targeted degradation22 of CDK4/6 might result in more potent inhibition of RB–E2F output and growth suppression of CDK4/6i-R tumor cells. As an approach, we developed hetero-bifunctional small molecules37 that would both inhibit CDK4/6 kinase activity as well as target CDK4/6 proteins for degradation. On the basis of the crystal structure of CDK6 in complex with palbociclib (Protein Database code 5L2I)38, we used solvent-exposed piperazine as the linker attachment point. We synthesized hetero-bifunctional potential CDK4/6-directed PROTACs by linking palbociclib to pomalidomide, a moiety with high affinity for the E3 ligase cereblon (CRBN), a component of a cullin-RING ubiquitin ligase complex39,40,41,42,43, using linkers of various lengths and types. By screening and optimizing synthesized compounds for their ability to degrade CDK4/6 and inhibit RB–E2F output in cells, we identified MS140 (Fig. 4a), as a highly potent CDK4/6 kinase inhibitor in vitro (Extended Data Fig. 4a) that markedly suppressed RB–E2F signaling and reduced CDK4 and CDK6 protein levels in a concentration and time-dependent manner (Fig. 4b,c). Degradation of CDK4/6 proteins by MS140 was specific, as shown by its abrogation upon pretreatment with excess palbociclib or pomalidomide (Fig. 4d and Extended Data Fig. 4b). We also confirmed that CDK4/6 protein degradation by MS140 was specifically mediated by the proteasome by its abrogation upon pretreatment with the proteasome inhibitor bortezomib or with the Nedd8-activating enzyme inhibitor MLN4924 (Fig. 4d and Extended Data Fig. 4b). Further, MS140 failed to degrade CDK4 in cells in which CRBN had been knocked out using CRISPR/Cas9 technology (Fig. 4e), confirming that target degradation by MS140 was specifically mediated by CRBN. As a negative control, we designed and synthesized MS140-ve (Extended Data Fig. 4c), a methyl analog of MS140 that is predicted not to bind CRBN42,44. Treatment with MS140-ve did not reduce CDK4/6 protein levels (Fig. 4f), further confirming that MS140 degrades its targets by linking them to the ubiquitin–proteasome machinery. Although palbociclib has been reported to bind a number of kinases in addition to CDK4/6, including CDK9 (ref. 45), we observed that MS140 did not degrade CDK7 or CDK9 (Fig. 4b,f). Finally, global analysis of protein degradation using mass spectrometry (MS) revealed an impressively selective protein downregulation profile of MS140, with very few hits other than CDK4 and CDK6 (Fig. 4g). Thus, MS140 potently and selectively both inhibited and degraded CDK4/6 kinases in CDK4/6i-S tumor cells by targeting them to the CRL4–CRBN–E3 ubiquitin complex.

a, Chemical structure of the bifunctional CDK4/6i-degrader MS140. b, Colo205 cells were treated with increasing concentrations of MS140 for 5 h. Lysates were immunoblotted with the indicated antibodies. c, Colo205 cells were treated with MS140 (0.5 µM) for the indicated time points. Lysates were immunoblotted with the indicated antibodies. d, Colo205 cells were pretreated with either the proteasome inhibitor 100 nM bortezomib (BOR), 10 µM PB, 10 µM pomalidomide (POM) or 1 µM MLN4924 (MLN) for 4 h, followed by treatment with MS140 (100 nM 3 h−1). Lysates were subjected to immunoblotting with the indicated antibodies. e, ZR-75-1 WT and CRBN-deficient cells were treated with the indicated concentrations of MS140 for 5 h. Lysates were immunoblotted with the indicated antibodies. f, Colo205 cells were treated with dimethyl sulfoxide (DMSO), MS140-ve and MS140 for 5 h. Lysates were immunoblotted with the indicated antibodies. g, Volcano plot of the protein log2 ratios representing the quantitative dynamics of 4,822 proteins in MS140 and MS140-ve (negative control of MS140) treated Colo205 samples (0.3 µM, 5 h) in duplicate. All data represent single experiments.
CDK4/6-directed degradation is more effective than CDK4/6is in CDK4/6i-S tumor cells
Treatment of various RB-proficient tumor cell lines with MS140 resulted in 3–30-fold greater suppression of both RB–E2F output and cell growth, compared to palbociclib, in the CDK4/6i-S cells identified in our first screen (Fig. 5a–d). A notable exception was H358, in which MS140 only modestly decreased CDK4 levels or inhibited RB signaling as compared to palbociclib (Fig. 5e). However, this cell line exhibited very low endogenous levels of CRBN (Fig. 1e), consistent with the increased effectiveness of MS140 being CRBN-dependent. We next assayed additional cell lines of hematopoietic origin. All mantle cell lymphoma (MCL) lines tested showed sensitivity to palbociclib and higher sensitivity to MS140 (Fig. 5f), associated with more potent and durable inhibition of RB–E2F output by MS140 (Fig. 5g and Extended Data Fig. 5a), consistent with these cells being predominantly CDK4-driven and expressing low levels of CDK6.

a, The indicated cell lines were subjected to cell growth crystal violet assay in the presence of PB or MS140 for 10–15 d. b, Colo205 cells were treated with increasing concentrations of PB or MS140 for 24 h. Lysates were immunoblotted with the indicated antibodies. c, The indicated cell lines were treated with increasing concentrations of PB or MS140 for 24 h. Lysates were immunoblotted with the indicated antibodies. d, U87MG or MCF7 cells were treated with increasing concentrations of PB or MS140 for 24 h. Lysates were immunoblotted with the indicated antibodies. e, H358 cells were treated with increasing concentrations of PB or MS140 for 24 h. Lysates were immunoblotted with the indicated antibodies. f, MCL cells were treated with PB or MS140 for 72–96 h. Cell viability was assayed using 0.1 mg ml−1 resazurin solution. Half maximal inhibitory concentration (IC50) values were determined by nonlinear regression curve fit in GraphPad Prism (n = 6 independent experiments). Data are presented as mean ± s.d. g, MCL cell lines were treated with increasing concentrations of PB or MS140 for 24 h. Lysates were immunoblotted with the indicated antibodies. h, KMS-12-PE or Pfeiffer cells were treated with increasing concentrations of PB or MS140 for 24 h. Lysates were immunoblotted with the indicated antibodies. i. The indicated cells were treated with PB or MS140 for 72–96 h. Cell viability was assayed using 0.1 mg ml−1 resazurin solution. IC50 values were determined by nonlinear regression curve fit in GraphPad Prism (n = 6 independent experiments). Data are presented as mean ± s.d. j, Mice carrying JeKo-1 xenografts were treated with vehicle (Veh) or MS140 (25 mg kg−1 twice daily) or PB (50 mg kg−1 once daily) for 3 d. Tumor samples were lysed and immunoblotted with the indicated antibodies. k, Mice carrying Colo205 xenografts were treated with vehicle or MS140 (30 mg kg−1, twice daily) for 3 d. Tumor samples were lysed and immunoblotted with the indicated antibodies. l, Scatter-plot of fold change for an efficacy assay in JeKo-1 tumor xenografts in mice treated with vehicle or MS140 (25 mg kg−1 twice daily) or PB (50 mg kg−1 once daily) for 21 d. Each treatment contained eight animals. Data are presented as mean ± s.e.m. Statistical significance was determined by paired two-tailed Student’s t-test. m, Scatter-plot of fold change for an efficacy assay in Colo205 tumor xenografts in nude mice treated with vehicle or MS140 (30 mg kg−1 twice daily) for 21 d. Vehicle treatment contained five animals. MS140 treatment contained eight animals. Data are presented as mean ± s.d. Statistical significance was determined by unpaired two-tailed Student’s t-test. n, qPCR analysis of expression of known RB–E2F target genes in tumors, kidney and liver from mice bearing JeKo-1 tumors treated with vehicle or MS140 (25 mg kg−1 twice daily) or PB (50 mg kg−1 once daily) for 3 d. Data are presented as mean ± s.d. (tumor, n = 1 mouse with three independent experiments; kidney and liver, n = 2 mice with two independent experiments for each mouse). o, Neutrophil counts in C57BL/6 mice before and after treatment with PB (50 mg kg−1 once daily, n = 8 mice) or MS140 (25 mg kg−1 twice daily, n = 7 mice) for 21 d. Data are presented as mean ± s.d. Statistical significance was determined by paired two-tailed Student’s t-test. All data represent single experiments. UD, undetected.
Of note, a smaller group of cancer cell lines expressing high levels of CDK6 compared to CDK4 are sensitive to CDK4/6is and highly sensitive to MS140 (Fig. 5h,i). These are mostly cells from hematopoietic origin, but also include the colorectal cancer cell line Colo205. These tumor cells dependent on CDK6 (refs. 12,29) (Extended Data Fig. 5b–d), indicating that in this subgroup of tumor cells, CDK6 is sensitive to CDK4/6is. Our data on growth response of hematopoietic tumor lines to palbociclib and MS140 are summarized in Extended Data Fig. 5e. Thus, MS140 was more effective than palbociclib in inhibiting RB–E2F output and cell growth of many CDK4/6i-S tumor cell lines. CDK4/6i-S cells are predominantly those expressing CDK4 and low CDK6, but we also identified a smaller subset of CDK6-dependent tumor cells that are also sensitive to CDK4/6is and more sensitive to the CDK4/6 degrader.
The increased effectiveness of MS140, as compared to palbociclib in CDK4/6i-S cells, prompted us to assess its biochemical and antitumor effects in vivo. Treatment of mice with MS140 promoted degradation of CDK4/6 kinases and suppression of RB–E2F output in JeKo-1 (MCL) and Colo25 (colorectal carcinoma) xenografts in vivo (Fig. 5j,k), resulting in equivalent or greater suppression of tumor growth compared to palbociclib administered at the same total daily concentration (Fig. 5l,m and Extended Data Fig. 5f) without significant body weight loss (Extended Data Fig. 5g) or other apparent toxicities.
The increased potency of MS140 in tumor cells compared to palbociclib, prompted us to assess the effect of MS140 on normal tissue to obtain some evidence on its potential therapeutic index. To this end, we determined the effect of treatment of the same daily dose of either MS140 or palbociclib on expression of known E2F targets in tumor and normal tissue (kidney and liver). In fact, the analysis revealed that treatment with MS140 resulted in more potent inhibition in the tumor than in normal tissue of several E2F-target transcripts, compared to palbociclib (Fig. 5n), suggesting that the strategy of combined inhibition and degradation of CDK4/6 may show higher therapeutic index than current clinical CDK4/6is. Finally, as a known toxicity induced by CDK4/6is is a reduction on white blood cells and neutropenia4, we performed complete blood counts in mice after 3 weeks of treatment with either palbociclib or MS140 at the same total daily concentration. Consistent with the clinical experience4 and previous studies in mice46, treatment with palbociclib resulted in a severe (>50%) reduction in neutrophil count (Fig. 5o). Total white blood cell count and lymphocytes were also reduced in response to palbociclib treatment, whereas red blood cells were slightly reduced (Extended Data Fig. 5h). However, neither the white blood cell nor lymphocytes count was decreased after treatment with MS140 (Fig. 5o), suggesting that the compound can be better tolerated at doses that are comparably effective with palbociclib. Thus, our results indicate that CDK4/6-directed degraders may be both a highly effective and well-tolerated therapeutic option for patients with CDK4-driven tumors, as well as for patients with tumors dependent on CDK4/6i-S CDK6.
In CDK4/6-R cells, CDK4/6 degraders fail to degrade CDK6 due to weak binding of compound
As MS140 both inhibits and degrades CDK4/6, we would expect it to show increased potency compared to the parent CDK4/6is in all cell lines. Notably however, in all CDK4/6i-R tumor lines tested, treatment with MS140 suppressed RB–E2F output and cell growth less effectively than palbociclib (Fig. 6a,b). This result was unexpected, as shRNA-mediated knockdown of CDK6 sensitized these same tumor cells to palbociclib (Fig. 3a,b), suggesting that MS140 downregulated CDK6 expression and activity with different potency in the two groups of cells lines. As MS140 is a bifunctional compound, the difference could be the result of either differential binding to CDK6 or difference in CRBN expression between the two groups of cell lines. As shown in Fig. 1e, with the exception of H358, CRBN is expressed at comparable levels in CDK4/6i-S and CDK4/6-R cell lines. Further, ectopic expression of CRBN in a CDK4/6i-R cell line did not increase CDK6 protein degradation or inhibition of downstream RB–E2F output by compound (Extended Data Fig. 6a). We thus concluded that differences in CRBN expression cannot be the underlying basis for the difference in sensitivity to MS140 and focused on the effect of MS140 directly on CDK6.

a, Cell growth crystal violet assay for the indicated cell lines treated with increasing concentrations of PB or MS140 for 10–12 d. b, A375, SKMEL2 and Calu6 cells were treated with increasing concentrations of PB or MS140 for 24 h. Lysates were immunoblotted with the indicated antibodies. c, A549 or A375 expressing Dox-inducible shCDK6 were treated with either the indicated concentrations of Dox (A375, 0.1 µg ml−1) or MS140 for 72 h. Lysates were subjected to immunoblotting with the indicated antibodies. d, Cell growth crystal violet assay for the indicated cell lines treated with either Dox or MS140 for 10 d. Colonies were stained with crystal violet. e, RB-proficient cell lines were treated with MS140 (3 nM) for the indicated time points. Cell lysates were immunoblotted with the indicated antibodies. f, The indicated cell lines were treated with PB (1 µM 2 h−1) followed by CETSA assay. The graphs are derived by immunoblot analysis of CDK6 and actin expression using ImageJ. Samples were derived from the same experiment and the blots were processed in parallel. g, The indicated cell lines were treated with MS140-ve (15 µM 2 h−1) followed by CETSA assay. The graphs are derived by immunoblot analysis of CDK6 and actin expression using ImageJ. The samples derive from the same experiment and the blots were processed in parallel. h, Desthiobiotin-ADP enrichment assay for CDK6 in MV4-11 and SKMEL2 cells pretreated with increasing concentrations of PB for 2 h. Elutes were subjected to immunoblotting with the indicated antibodies. All data represent single experiments.
We first compared the extent of reduction of CDK6 protein expression in response to shRNA-mediated CDK6 knockdown or MS140 treatment. As shown in Fig. 6c,d, MS140 was less potent in promoting CDK6 protein degradation and in suppressing cell growth, compared to shRNA-mediated CDK6 knockdown. These results prompted us to directly compare the extent of CDK4/6 degradation by MS140 in CDK4/6i-S and CDK4/6i-R cell lines. Treatment with MS140 resulted in potent degradation of CDK4/6 in both CDK4-driven and CDK6-driven CDK4/6i-S cells, but promoted only minimal CDK4/6 degradation in CDK6-driven CDK4/6i-R cells (Fig. 6e). To assess whether this difference applied more generally to CDK4/6 degraders, we tested two recently reported CDK4/6-directed PROTACs, YKL-06-102 (refs. 23,24) and BSJ-02-162 (ref. 24). We found that, similarly to MS140, treatment with either compound resulted in more potent degradation of CDK6 in CDK4/6i-S as compared to CDK4/6i-R cells (Extended Data Fig. 6b,c). Of note, one of these compounds has been reported to be a CDK6-selective inhibitor, compared to CDK4. We confirmed that, as reported, that compound was a relatively more potent degrader of CDK6 than CDK4 in CDK4/6-S cells, compared to other CDK4/6 degraders (data not shown). However, as shown in Extended Data Fig. 6b,c, YKL-06-102 was similarly ineffective to the rest of CDK4/6 degraders in degrading CDK6 in CDK4/6-R cells. Thus, failure to degrade CDK4/6 in CDK4/6i-R cells is a general property of CDK4/6 degraders.
To directly assess binding of CDK4/6is to CDK6 in cells, we compared the shift of the CDK6 melting temperature (Tm) promoted by CDK4/6is in CDK4/6i-S versus CDK4/6i-R tumor cells (all driven by CDK6) by cellular thermal shift assay (CETSA)47,48. Treatment of CDK4/6i-S, but not CDK4/6i-R, cells with palbociclib resulted in a shift in Tm, indicating strong inhibitor binding to CDK6 in these cells (Fig. 6f). Similar results were obtained with MS140-ve, used instead of MS140 to make sure there is no CRBN-mediated protein degradation that could confound the experiment (Fig. 6g). Finally, to assess inhibitor binding in cells using a second method, we employed a probe-based chemoproteomic method using biotinylated phosphates of ATP or ADP that irreversibly react with protein kinases on conserved lysine residues in the ATP binding pocket49, followed by western blot analysis. We found that palbociclib prevented binding of the probe in CDK4/6i-S cells but not in CDK4/6i-R tumor cells (Fig. 6h). Together these data show that CDK6-dependent tumor cells can be divided in two groups. In most tumor cells that express CDK6, CDK4/6is bind weakly to CDK6 and these cells are resistant to CDK4/6is (and consequently to CDK4/6 degraders). In a smaller group of CDK6-dependent cells, CDK6 binds strongly to CDK4/6is and they are thus sensitive to CDK4/6is and degraders.
CDK4/6i-R cells express CDK6 as a thermostable, weak HSP90 client protein
To gain mechanistic insight on the basis of the difference in binding of CDK6 to CDK4/6is, we carried out protein expression analysis of the main known CDK4/6-related regulators (Fig. 7a), as well as immunoprecipitation of CDK6 followed by MS analysis of interacting partners in a CDK4/6-S (KMS-12-PE) and in a CDK4/6i-R (Calu6) cell line (both expressing high levels of and dependent on CDK6) (Fig. 7b). Association of CDK6 with known CDK6 interactors, including members of the cyclin, INK4 or CIP/KIP families correlated well with their relative basal expression in the two cell lines (Fig. 7c and Extended Data Fig. 7a), suggesting that additional factors affect tumor cell sensitivity to CDK4/6is. For example, p27 was found to be expressed at high levels and associate strongly with CDK6 in a CDK4/6i-S cell line (KMS12PE) as well as in a CDK4/6i-R line (A375) (Fig. 7c). We however noticed a threefold to fivefold higher interaction of CDK6 with multiple components of the HSP90–CDC37 chaperoning complex in CDK4/6i-S compared to CDK4/6i-R cells (Fig. 7b and Extended Data Fig. 7a). The observation was further confirmed by direct co-immunoprecipitation experiments showing a much stronger interaction of CDK6 with HSP90 and CDC37 in CDK4/6i-S as compared to CDK4/6i-R tumor cells (Fig. 7d and Extended Data Fig. 7b).

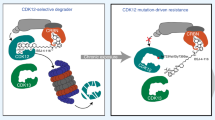
a, Immunoblots for cell cycle regulators in CDK6-dependent cell lines. LE, long exposure. b, Volcano plot of the CDK6-interacting proteins in KMS-12-PE and Calu6. Proteins in red were annotated as HSP90–CDC37-related. c, Cell lysates from the indicated cell lines were either subjected to Co-IP with a CDK6 antibody followed by immunoblotting with known cell cycle regulators or immunoblotted with the indicated antibodies. d, Cell lysates from the indicated cell lines were either subjected to Co-IP with a CDK6 antibody followed by immunoblotting with HSP90, CDC37 and CDK6 or immunoblotted with the indicated antibodies. e, Cell lines were treated with the HSP90 inhibitor ganetespib (GAN) (30 nM) at the indicated time points. Cell lysates were immunoblotted with the indicated antibodies. f, Immunoblots for CDK6 CETSA in CDK4/6i-S and CDK4/6i-R cell lines heat-treated at increasing temperature end points. g, The indicated cell lines were treated with 100 µg ml−1 cycloheximide (CHX) at the indicated time points. Cell lysates were immunoblotted with the indicated antibodies. h, A375 cells were transfected with either WT or CDK6 (S178P) followed by treatment with increasing concentrations of MS140 for 24 h. Cell lysates were immunoblotted with the indicated antibodies. i, KMS-12-PE and BT549 cells were treated with increasing concentrations of MS140 for 24 h. Lysates were subjected to immunoblotting with the indicated antibodies. j, Model of CDK6 association with the HSP90 complex affecting tumor cell sensitivity to CDK4/6is and degraders. In CDK4/6i and degrader-sensitive cells CDK6 is associated with the HSP90 complex (top). CDK4/6is or CDK6 degraders bind CDK6 strongly and promote CDK6 inhibition or both CDK6 inhibition and degradation, respectively. In CDK4/6is and degrader-resistant cells, CDK6 is weakly associated with the HSP90 complex (bottom). In these cells, CDK4/6is and degraders bind CDK6 weakly and thus fail to promote CDK6 inhibition or both inhibition and degradation, respectively. All data represent single experiments.
Association of a kinase with the HSP90–CDC37 complex has been shown to correlate with kinase dependence on HSP90 for its folding and stability15. In fact, treatment with the HSP90 inhibitor ganetespib resulted in much less potent CDK6 degradation in CDK4/6i-R cells, than in CDK4/6i-S cells (Fig. 7e and Extended Data Fig. 7c). A structurally distinct HSP90 inhibitor, luminespib showed the same results (Extended Data Fig. 7d). Moreover, CDK6 protein levels remained unchanged after siRNA-mediated knockdown of CDC37 in a CDK4/6-R line (Extended Data Fig. 7e), further confirming that the HSP90–CDC37 chaperoning activity is not required for CDK6 protein stability in these cells. As expected, treatment of CDK4-only-expressing cells with ganetespib resulted in potent degradation of CDK4 (Extended Data Fig. 7f), a known strong HSP90 client18,50. Other known HSP90 clients were degraded to a similar degree upon ganetespib treatment (Extended Data Fig. 7g), indicating that the HSP90–CDC37 chaperone system operates similarly in the two settings, but CDK6 in CDK4/6i-S cells is expressed as a strong HSP90 client, whereas in CDK4/6i-R cells it is predominantly expressed as a weak HSP90 client.
Strong HSP90 client kinases have been shown to be highly thermo unstable15. We compared the thermostability of CDK6 in CDK6-driven CDK4/6i-S versus CDK4/6i-R tumor cells using CETSA. Consistent with the relative association of the two CDK6 forms with HSP90, we found much lower thermostability (lower Tm) of CDK6 protein in CDK4/6i-S cells as compared to that of CDK6 expressed in CDK4/6i-R cells (Fig. 7f). CDK6 in the two cellular settings also differed remarkably in its overall stability. Cycloheximide treatment resulted in significantly faster degradation of CDK6 in CDK4/6i-S compared to CDK4/6i-R cells (Fig. 7g).
Stronger association of a kinase with HSP90 has been reported to shift the conformational ensemble of different activation states toward a more active distribution18,51. Crystallographic data show that CDK4/6is are type I, in that they bind the active conformation of CDK4/6 (αC-IN/DFG-IN)52. Our findings support a model by which, in CDK6-driven and CDK4/6i-S tumor cells, the CDK6 protein population is enriched in highly active conformations that have strong binding affinity for current CDK4/6is (and consequently for CDK4/6 degraders). In contrast, in CDK4/6i-R cells, CDK6 is expressed as a thermostable, weak HSP90 client, with lower affinity for CDK4/6is. To further test this idea, we inserted a previously reported activating mutation in CDK6 (S178P)53 and we confirmed that it activated CDK6 (Extended Data Fig. 7h). Treatment of cells expressing either WT or mutationally activated CDK6 with MS140 resulted in substantially more potent degradation of CDK6 (S178P) compared to CDK6 WT (Fig. 7h), consistent with a highly active conformation of CDK6 being more sensitive to degradation by MS140. Finally, a previous report using biotinylated phosphates of ATP or ADP followed by MS has shown that in RB-null cells, palbociclib does not bind CDK4/6 kinases54. Consistent with those results, we found that in BT549, RB-null cells, CDK6 is also highly thermostable and resistant to degradation by MS140, similar to the CDK6 state in palbociclib-resistant cells (Fig. 7i and data not shown).
Discussion
We show here that the expression state of CDK6 is a critical regulator of tumor response to CDK4/6is. Cells universally express CDK4, but CDK6 expression varies substantially across tumor types as well as within the same tumor type. We found that tumors with low CDK6 expression relative to CDK4 are sensitive to CDK4/6is. Such tumor types include luminal breast cancer, Ewing sarcomas, MCLs and others, as well as subgroups within tumor types, such as a portion of lung adenocarcinomas. Among lung adenocarcinoma models, we found a subgroup with a low ratio of CDK6 to CDK4 protein expression that was sensitive to CDK4/6is. The association of low CDK6 expression with increased clinical response to CDK4/6is was further confirmed by analyzing clinical data in patients with NSCLC27. Thus, collectively our data indicate that a low ratio of CDK6 to CDK4 expression in the tumor may be used to stratify patients who are likely to benefit from CDK4/6-based therapies. Further, it is becoming increasingly apparent that in various contexts of intrinsic or acquired resistance, combinatorial targeting of parallel or convergent growth factor signaling pathways, such as the PI3K–PTEN–AKT55,56,57 or RAS–MAPK25,58 pathway may be necessary for effective and durable tumor response. Our findings suggest that selection of patients with low CDK6 relative to CDK4 expression, in combinations of CDK4/6is or CDK4/6 degraders with inhibitors of parallel pathways, such as PI3K, AKT, MEK or ERK inhibitors are likely to be most effective.
We further found that in most tumor cells, CDK6 expression confers intrinsic resistance to CDK4/6is. The finding is consistent with CDK6 overexpression being a reported mechanism of acquired resistance to CDK4/6is in luminal breast cancer13,59. However, CDK4/6is have been shown here (Extended Data Fig. 4a) and by others1 to potently bind purified CDK6 in vitro. In addition, a proportion of tumors (mostly of certain types of leukemias, such as acute myeloid leukemia) have been shown to require CDK6 (ref. 14) and are sensitive to CDK4/6is, suggesting that CDK6 may or may not confer resistance to CDK4/6is depending on cellular context. We used a potent and selective CDK4/6 degrader that we developed (MS140) as a tool to resolve the discrepancy between the in vitro data and our findings in cells. By employing in-cell binding assays, we found that CDK6 is expressed in different tumors in either of two distinct forms, only one of which is accessible to binding by CDK4/6is (Fig. 7h). We further found that the two CDK6 forms differ dramatically in their thermostability and their dependence on the HSP90–CDC37 chaperone complex. The data are consistent with previous studies showing kinase dependence on HSP90 to be associated with an enrichment of folded, highly active states of the kinase18, which, in the case of CDK6, is structurally predicted to bind strongly to CDK4/6is. The conformation of the active site of the highly active CDK6 in cells likely resembles that of the highly active purified CDK6, commonly used in in vitro kinase and binding assays. Thus, the current clinical CDK4/6is have been developed to strongly bind and inhibit CDK6 selectively in the subgroup of tumor cells that express CDK6 similar to its purified, highly active state.
Recently, two studies have proposed the interaction of CDK4 to either p27 (ref. 60) or p16 (ref. 61) as mechanisms promoting resistance to CDK4/6is. In another study, formation of complexes with members of the cyclin D family or with endogenous inhibitors of CDKs was shown to affect the interaction of different members of the CDK family of proteins with inhibitors and HSP90–CDC37 in vitro21. However, we observed p16 to be expressed at undetectable levels in most cancer cell lines tested in our experiments, including the ones that are insensitive to CDK4/6is (Figs. 1e and 7a) and we did not observe a correlation of endogenous expression of members of the cyclin D family with either CDK6 sensitivity to CDK4/6is or HSP90 dependence. Further, p27 is expressed in most CDK4/6i-S and CDK4/6-R cells at similar levels (Figs. 1e and 7a) and knockdown or overexpression of p27 did not affect sensitivity of cell lines to CDK4/6is (data not shown). Finally, we found that all cell lines tested expressing CDK4 and low levels of CDK6 are universally sensitive to CDK4/6is, regardless of p16 or p27 status or expression levels. We therefore conclude that although the reported mechanisms may affect target engagement by CDK4/6is or the interaction of CDK4 or CDK6 with HSP90–CDC37 in certain contexts, they are unlikely to be the predominant mechanism by which large subsets of RB-proficient tumors diverge in terms of their sensitivity to CDK4/6is.
The duality of CDK6 when expressed in different cells with respect to its dependence on HSP90–CDC37 challenges the current paradigm on the molecular determinants of kinase dependence on HSP90. HSP90–CDC37 dependency of a given kinase has been shown to be altered by genetic alterations, as in the cases of c-Src and v-Src15, WT and mutated BRAF19 and WT and mutated EGFR20. Previous studies using ectopically expressed proteins, have shown that CDK6 exhibited an intermediate dependency on HSP90 (HSP90 interaction score of 2.23), lying in between the highly dependent CDK4 (score of 3.15) and the entirely HSP90-independent CDK2 (score of 0)15. However, we unexpectedly found that the same kinase (CDK6) can be expressed as a strong or weak HSP90–CDC37 client depending on the cellular context. As regulation of kinase function by the HSP90–CDC37 complex remains incompletely understood15, in the case of CDK6 may be affected by post-translational modification(s), interacting partner(s) or localization, in different cellular environments.
Current clinical CDK4/6is have been optimized to bind and inhibit CDK4 as well as the thermo-unstable, strong HSP90 client form of CDK6. However, based on our data, it is expected that a large number of tumors express thermostable CDK6, which is resistant to these drugs. Drug development efforts directed in potently targeting this newly identified state of CDK6 may bring about more effective therapies for a large number of patients with RB-proficient tumors.
Methods
Compounds
Commercially available compounds palbociclib, ganetespib, luminespib and bortezomib were obtained from Selleckchem. Abemaciclib, ribociclib and MLN4924 were obtained from Medchem Express. Blasticidin S, CHX and DOX were purchased from Sigma. Chemical compounds were dissolved either in DMSO or water and stored at −20 °C.
Cell culture
Cell lines HEK293T, HEK293FT, Huh-7, U87MG, A375 and A673 were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mM glutamine and penicillin/streptomycin, MCF7, T47D, ZR-75-1, CAMA-1, NCI-H358, Colo205, SK-MEL-1, IGROV-1, TC-71, Mino, JeKo-1, Granta 519, Z-138, REC-1, KMS-12-PE, MV4-11, MOLM-14, Pfeiffer, SK-MEL-2, A549, Calu6, NCI-H1792, NCI-H2087, NCI-H2291, NCI-H1915, NCI-H1666, NCI-H1395, HCC827, PC9, HCT15, HCT116, RKO, LoVo, HT-29, SW620 and BT549 were maintained in RPMI1640 supplemented with 10% fetal bovine serum, 2 mM glutamine and penicillin/streptomycin (Thermo Fisher Scientific).
MCF7, T47D, ZR-75-1, CAMA-1, NCI-H358, Colo205, A375, A549, SK-MEL-2, Calu6, NCI-H1792, NCI-H2087, NCI-H2291, NCI-H1915, NCI-H1666, NCI-H1395, HCC827, RKO, HCT15, HCT116, SW620, LoVo, HT-29 and HEK293T were purchased from the American Type Culture Collection. PC9 cells were purchased from Sigma-Aldrich. U87MG, IGROV-1 and BT549 were provided by R. Parsons. Huh-7 cells were provided by A. Lujambio. Z-138 cells were provided by E. P. Reddy. REC-1, KMS-12-PE and Pfeiffer were gifts from S. Parekh. MV4-11 and MOLM-14 cells were provided by I. Aifantis (New York Univeristy). Mino, JeKo-1 and Granta 519 were gifts from S. M. Buckley (University of Nebraska). A673 and TC-71 were provided by C. A. Pratilas (John Hopkins). HEK293FT cells were a gift from W. Kaelin Jr (Harvard Medical School).
Plasmids
Tet-pLKO-puro (cat. no. 21915) and pcDNA3-FLAG-CRBN (cat. no. 107380) was purchased from Addgene. shCDK4 (TRCN0000018364 and TRCN0000010520), shCDK6 (TRCN0000010081 and TRCN0000000488) and shCDC37 (TRCN0000116633 and TRCN0000116632) (Supplementary Table 1) were subcloned into Tet-pLKO plasmid. Human CDK6 coding sequences were amplified from HEK293H cDNA using a SuperScript III First-Strand kit (Thermo Fisher Scientific). C-terminal V5-tagged CDK6 were subcloned using pcDNA3 as a backbone. shRNA-resistant form of V5-CDK6 and S178P V5-CDK6 were generated with a QuikChange II XL Site-Directed Mutagenesis kit (Agilent).
siRNA knockdown
A673, TC-71 and NSCLC cell lines were transfected with SMARTpool ON-TARGETplus Non-targeting pool or SMARTpool ON-TARGETplus CDK4-siRNA (L-003238-00-0005), or SMARTpool ON-TARGETplus CDK6-siRNA (L-003240-00-0005) (Dharmacon) using Lipofectamine RNAiMAX (Thermo Fisher Scientific).
Western blot and immunoprecipitation
Cells were washed with PBS and lysed on ice for 10 min in NP40 buffer (50 mM Tris (pH 7.5), 1% NP40, 150 mM NaCl, 10% glycerol, 1 mM EDTA) supplemented with protease and phosphatase inhibitors (Roche). Lysates were centrifuged at 15,000 r.p.m. for 10 min and the protein concentration was quantified using BCA (Thermo Fisher Scientific). Protein G agarose (Thermo Fisher Scientific) was used for immunoprecipitations. The following antibodies were used: phospho-RB (Ser807/811, 1:1,000 dilution), RB (4H1, 1:1,000 dilution), PLK1 (208G4, 1:1,000 dilution), cyclin D2 (D52F9, 1:1,000 dilution), cyclin E2 (1:1,000 dilution), Akt1 (2H10, 1:1,000 dilution), JAK2 (D2E12, 1:1,000 dilution), DHFR, ß-actin (13E5, 1:1000), anti-rabbit, HRP (1:1000) and anti-mouse, HRP (1:1000) (Cell Signaling), CDK4 (H-22), CDK6 (C-21; DCS-83, 1:1,000 dilution), CDK2 (D-12, 1:1,000 dilution), CDK7 (C-19, 1:2,000 dilution), CDK9 (D-7, 1:3,000 dilution), p15/16 (C-7, 1:400 dilution), p16 (C-20, 1:500 dilution), p18 (118.2, 1:250 dilution), p57 (KP39, 1:500 dilution), cyclin A (H-432, 1:2,000 dilution; B-8, 1:1,000 dilution), cyclin D1 (M-20, 1:400 dilution; A-12, 1:250 dilution), cyclin D3 (D-7, 1:500 dilution), cyclin E1 (HE12, 1:2,000 dilution), CDC37 (H-271; E-4, 1:2,000 dilution), Hsp70 (W27, 1:8,000 dilution) and Hsp90α/ß (F-8, 1:8,000 dilution) (Santa Cruz), p27 (1:1,000 dilution) and c-Raf (1:2,000 dilution) (BD Transduction Laboratories), p21 (1:1,000 dilution) and phospho-CDK4-T172 (1:1,000 dilution) (ABclonal), CRBN (1:500 dilution; Novus Biologicals), V5 (1:5,000 dilution; Thermo Fisher Scientific) and FLAG M2 (1:3,000 dilution; Sigma).
In vitro CDK4 and CDK6 kinase assay
Palbociclib, MS140 and ribociclib were assayed by Reaction Biology Corporation for in vitro kinase activity. Briefly, kinase substrates were added to base reaction buffer (20 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg ml−1 BSA, 0.1 mM Na3VO4, 2 mM dithiothreitol and 1% DMSO). Differential CDK4/6 complexed with cyclin D1 were then diluted in the substrate solution. A range of concentrations of compounds were incubated with the kinase reaction mixture by acoustic technology (Echo550; nanoliter range) and incubated for 20 min at room temperature followed by the addition of 10 μM 33P-ATP. Reactions were incubated for 2 h at room temperature and radioactivity was detected by filter binding methods. Kinase activity data were presented as percentage kinase activity in samples relative to DMSO control. IC50 values were calculated using GraphPad Prism 5.
Lentivirus transduction and stable cell lines
Lentivirus was produced by transfecting HEK293FT cells with a lentiviral transfer vector, psPAX2 and pMD.G at a 5:4:1 ratio using Lipofectamine 2000 (Thermo Fisher Scientific). The viral supernatant was collected 72 h after transfection and filtered through a 0.45-µm filter unit (Millipore). For Dox-inducible stable CDK4/6 knockdown cell lines, cells were transduced with shCDK4 or shCDK6 lentivirus in the presence of 8 µg ml−1 polybrene (EMD Millipore) and selected with 2 µg ml−1 puromycin (Thermo Fisher Scientific) for 5–7 d.
Cellular thermal shift assay
For basal CDK6 thermal stability assay, cells resuspended in 1 ml PBS containing protease inhibitor cocktail (Roche) were aliquoted with 100 µl cell suspension each into nine PCR tubes (~3 × 106 cells). PCR tubes were heated at different temperature end points (35–59 °C) for 3 min in the Veriti 96-well thermal cycler (Thermo Fisher Scientific). Tubes were removed and incubated at room temperature for 3 min. Samples were immediately snap-frozen in liquid nitrogen. Cells were frozen and thawed three times using liquid nitrogen and a thermal cycler at 25 °C. Cell lysates were centrifuged at 20,000g for 15 min at 4 °C. Supernatants were collected for western blots.
For palbociclib or MS140-ve-induced thermal shift assay, cells were treated with 1 µM palbociclib or 15 µM MS140-ve for 2 h. Cells resuspended in PBS containing protease inhibitor cocktail (Roche) were aliquoted with 100 µl cell suspension each into nine PCR tubes. Samples were heated at different temperature end points for 3 min using the Veriti 96-well thermal cycler (Thermo Fisher Scientific), followed by incubation at room temperature for 3 min and snap-freezing in liquid nitrogen. After three freeze–thaw cycles, samples were centrifuged at 20,000g for 15 min at 4 °C. Supernatants were collected for western blot analysis.
ATP-biotin competition assay
Cell lysates were prepared and labeled according to the manual instructions for the Pierce Kinase Enrichment Kits and ActivX Probes (Thermo Fisher Scientific). Briefly, cells were pretreated with palbociclib for 2 h, lysed and centrifuged at 16,000g for 10 min at 4 °C. The supernatant was desalted through Zeba Spin Desalting Columns. Then, 1 mg of total cell lysates in 500 µl were used for ATP competition reaction with a final concentration of 5 µM desthiobiotin-ADP probe for 10 min at room temperature. Samples were mixed with 500 µl 8 M urea and 50 µl streptavidin agarose for 1 h at room temperature on a rotator. Beads were washed with 4 M urea/lysis buffer and collected by centrifugation at 1,000g for 1 min. Proteins were eluted with 2× sample buffer at 95 °C for 5 min. Samples were analyzed by immunoblotting.
Generation of CRBN −/− ZR-75-1 cell line by CRISPR/Cas9
Human CRBN gRNA (5′-CACCGTAAACAGACATGGCCGGCGA-3′) was designed using the following website: http://crispr.mit.edu/ and cloned into BsmBI-digested lentiCRISPRv2 (Addgene, 52961). ZR-75-1 cells were transduced with sgCRBN lentivirus in the presence of 8 µg ml−1 polybrene followed by selecting with 2 µg ml−1 puromycin for 5 d. Cells were plated at 0.3 cells per well in a 96-well plate. After 2–3 weeks, individual clones were expanded. CRBN homozygous knockout clones were validated by genotyping with primers (F: 5′- AAG TCA TGC TAA GGG CTG GAA C -3′, R: 5′- GGA TGG GTT TCC TGT TCT TAA TAG -3′) and western blotting.
Protein extraction and digestion
Colo205 cells were treated with 0.3 µM MS140 or MS140-ve for 5 h in duplicate. Cell pellets were lysed in lysis buffer (8 M urea, 100 mM Tris-HCl pH 8.0). Sonication (5 s on, 5 s off; 2 × 30 s) was performed to shear genome DNA. Lysates were centrifuged for 30 min at 3,500g at 4 °C, and the supernatants were transferred to clean tubes. Protein concentration was determined (BCA assay), and protein was reduced with 5 mM dithiothreitol (DTT), alkylated with 15 mM iodoacetamide (IAA) in the dark, then diluted with 3× volume of 25 mM Tris (pH 8.0), 1 mM CaCl2. The final urea concentration was 2 M. Trypsin was added into the protein solution with 1:100 ratio (trypsin:protein) and digested for 12–16 h or overnight at room temperature.
Mass spectrometry analysis
Peptides were cleaned up by C18 stage tips, and peptide concentration was determined (Peptide assay, Thermo 23275). The clean peptides were dissolved in 0.1% formic acid and analyzed on a Q-Exactive HF-X coupled with an Easy nanoLC 1200 (Thermo Fisher Scientific). 0.5 µg of peptides was loaded onto an Acclain PepMap RSLC C18 column (150 mm × 75 μm inner diameter (ID), C18, 2 μm, Thermo Fisher). Analytical separation of all peptides was achieved with 100 min gradient. A linear gradient of 5 to 30% buffer B over 80 min was executed at a 300 nl/min flow rate following by a ramp to 100% buffer B in 5 min, and a 15-min wash with 100% buffer B, where buffer A was aqueous 0.1% formic acid and buffer B was 80% acetonitrile and 0.1% formic acid. LC-MS experiments were performed in a data-dependent mode with full MS (externally calibrated to a mass accuracy of <5 p.p.m. and a resolution of 60,000 at m/z 200) followed by high-energy collision-activated dissociation-MS/MS of the top 20 most intense ions with a resolution of 15,000 at m/z 200. High-energy collision-activated dissociation-MS/MS was used to dissociate peptides at a normalized collision energy of 27 eV in the presence of nitrogen bath gas atoms. Dynamic exclusion was 30.0 s. There were two biological replicates for one treatment, and each sample was subjected to two technical LC-MS replicates.
MS data analysis
Mass spectra processing and peptide identification were performed on the Andromeda search engine in MaxQuant software (version 1.6.10.43) against the human UniProt database (UP000005640). All searches were conducted with a defined modification of cysteine carbamidomethylation, with methionine oxidation and protein amino-terminal acetylation as dynamic modifications. Peptides were confidently identified using a target-decoy approach with a peptide false discovery rate (FDR) of 1% and a protein FDR of 1%. A minimum peptide length of seven amino acids was required, maximally two missed cleavages were allowed, initial mass deviation for precursor ion was up to 7 p.p.m., and the maximum allowed mass deviation for fragment ions was 0.5 Da. Data processing and statistical analysis were performed on Perseus (version 1.6.10.50). Protein quantitation was performed on biological and technical replicate runs, and two-sample t-test statistics was used to report statistically significant expression fold changes.
MS to identify CDK6-interacting proteins
Preparation of samples for MS
The affinity purified proteins were reduced, alkylated and digested with trypsin directly on beads. Briefly, beads were resuspended in 100 μl 100 mM ammonium bicarbonate. Proteins were reduced with 2 μl of 0.2 M dithiothreitol (Sigma) for 1 h at 57 °C (pH 7.5), alkylated with 2 μl of 0.5 M iodoacetamide (Sigma) for 45 min at room temperature (RT) in the dark and digested using 200 ng sequencing-grade trypsin (Promega) overnight at RT with gentle shaking. The solution was transferred to a new tube and digestion was stopped by adding 100 μl of a 5% formic acid and 0.2% TFA R2 50 μm Poros (Applied Biosystems) beads slurry in water. The samples were allowed to shake at 4 °C for 3 h. Beads were loaded onto C18 ziptips (Millipore), equilibrated with 0.1% TFA, using a microcentrifuge for 30 s at 6,000 r.p.m. Beads were washed with 0.5% acetic acid. Peptides were eluted with 40% acetonitrile in 0.5% acetic acid followed by 80% acetonitrile in 0.5% acetic acid. Organic solvent was removed using a SpeedVac concentrator and the sample was reconstituted in 0.5% acetic acid.
MS analysis
One-tenth of each sample was loaded onto an Acclaim PepMap trap column (2 cm × 75 µm) in line with an EASY-Spray analytical column (50 cm × 75 µm ID PepMap C18, 2-μm bead size) using the auto sampler of an EASY-nLC 1200 HPLC (Thermo Fisher Scientific) with solvent A consisting of 2% acetonitrile in 0.5% acetic acid, and solvent B consisting of 80% acetonitrile in 0.5% acetic acid. Peptides were gradient-eluted into a Thermo Fisher Scientific Q Exactive HF-X Mass Spectrometer using the following gradient: 5–35% in 60 min, 35–45% in 10 min, followed by 45–100% in 10 min. High-resolution full MS spectra were recorded with a resolution of 45,000, an AGC target of 3×106, with a maximum ion time of 45 ms and a scan range from 400 to 1,500 m/z. MS/MS spectra were collected using a resolution of 15,000, an AGC target of 1×105, maximum ion time of 120 ms, one microscan, 2-m/z isolation window and normalized collision energy of 27.
Data processing
MS/MS spectra were searched against the UniProt human reference proteome database containing common contaminant proteins using Sequest within Proteome Discoverer 2.3. Search parameters were as follows: precursor mass tolerance ±10 ppm, fragment mass tolerance ±0.02 Da, digestion parameters trypsin allowing two missed cleavages, fixed modification of carbamidomethyl on cysteine, variable modification of oxidation on methionine, variable modification of deamidation on glutamine and asparagine and a 1% peptide and protein FDR searched against a decoy database. The results were filtered to only include proteins identified by at least two unique peptides.
Cell viability assay
Cells were seeded at 3–10 × 103 cells per well in 96-well plates. At 24 h after seeding, cells were treated with palbociclib and MS140 at a range of concentrations for 72 h. Ten microliters of 0.1 mg ml−1 resazurin (Sigma-Aldrich) or 20 µl of CellTiter 96 AQueous One Solution Reagent (Promega) was added to cells and incubated for 2–3 h at 37 °C. Cell viability was determined by measuring the fluorescence at 560 nm excitation wavelength and 590 nm emission wavelength (resazurin) or 490 nm (CellTiter 96 AQueous One Solution Reagent) using a Molecular Devices Spectramax M5 plate reader. IC50 values were calculated using log-transformed, normalized data in GraphPad Prism 5.0.
Crystal violet cell growth assay
Cells were seeded at 1–10 × 103 cells per well in six-well plates. The next day, cells were treated with the increasing concentrations of palbociclib and MS140 for 10–15 d. Cell culture medium was replaced every 2 d in the presence or absence of inhibitors. Cells were fixed with 10% formalin solution (Sigma-Aldrich) for 5 min at RT followed by 0.05% crystal violet for 25 min. cells were de-stained with tap water and air dried.
Quantitative real-time PCR
Total RNA was extracted using Trizol Reagent (Thermo Fisher Scientific). Complementary DNA was synthesized with a SuperScript IV First-Strand kit (Thermo Fisher Scientific). Quantitative real-time PCR was performed using a Fast SYBR Green Master Mix kit (Thermo Fisher Scientific) with a 7500 Fast real-time PCR system (Applied Biosystems). PCR primers are listed in Supplementary Table 1. Differences in expression were calculated by the ΔΔCt method.
Genetic dependency data and genomics data
CRISPR dependency data were obtained from the 20Q1 public Avana dataset containing genome-scale CRISPR knockout screens for 18,333 genes in 739 cell lines. The gene dependencies were estimated for each gene and cell lines by the CERES algorithm31. RNA interference (RNAi) dependency data were derived from combination of the Broad Institute Project Achilles, Novartis Project Drive and Marcotte et al. database32,33,34,35. Genetic dependencies were estimated using the DEMETER2 model across 712 unique cancer cell lines36. Cancer cell line mRNA expression was taken from the DepMap 20Q1 data release. Cancer Cell Line Encyclopedia proteomics data were obtained by quantitative profiling of proteins by MS across 375 cell lines1. Normalized protein quantification data and cell line omics data can be downloaded at DepMap (https://depmap.org/portal/).
Animal experiments
To determine in vivo degradation efficacy, 5–7-week-old female athymic Nude-Foxn1nu mice obtained from Envigo Laboratories were injected subcutaneously with 1 × 107 JeKo-1 cells in 1:1 PBS/Matrigel GFR membrane matrix (Corning) or 5 × 106 Colo205 cells in PBS. Mice were treated with vehicle (5% DMSO and 95% PEG 300) or MS140 (25 or 30 mg kg−1) intraperitoneally, twice daily or palbociclib (50 or 60 mg kg−1) orally once daily for 3 d when tumors reach around 100 mm3. At 5 h after the last dose, tumors were collected for further analysis. Liver and kidney were collected for qPCR analysis.
For efficacy of MS140 on tumor xenograft, 5 × 106 Colo205 cells in PBS or 10 × 106 JeKo-1 cells in 1:1 PBS/Matrigel GFR membrane matrix were injected subcutaneously on the left flank in 6-week-old female athymic Nude-Foxn1nu mice. Tumors were allowed to reach 100 mm3 in size before the animals were randomized into two groups of 5–8 mice per group. Mice were treated with vehicle (5% DMSO and 95% PEG 300) or MS140 (25 or 30 mg kg−1) intraperitoneally, twice daily or palbociclib (50 or 60 mg kg−1) orally once daily for 3 weeks. Tumor size was measured using calipers every 3 d and tumor volume was calculated using the following formula: (length × width2) / 2. Tumor samples and organs were collected at the end of treatment for further analysis. All experiments were conducted under a protocol approved by Mount Sinai School of Medicine Institutional Animal Care and Use Committee (IACUC-2016-0066). All mice were maintained in specific-pathogen-free barrier conditions under a 12-h-light–12-h-dark cycle and with temperature and humidity set points at 20–25% and 30–70%, respectively.
Complete blood count
Female 5–7-week-old C57BL/6 mice (Envigo Laboratories) were dosed intraperitoneally with 25 mg kg−1 MS140 twice daily or 50 mg kg−1 palbociclib once daily via oral gavage for 3 weeks. Mouse blood samples were collected in K3EDTA tubes before treatment and last treatment. Complete blood counts were performed with a Coulter Ac•T 5diff Hematology Analyzer (BECKMAN).
Trial design
The JUNIPER study was a randomized phase III study of abemaciclib with best supportive care versus erlotinib with best supportive care in patients with stage IV NSCLC with a detectable KRAS mutation whose disease had progressed after platinum-based chemotherapy.
RNA sequence
A total of 148 patients had evaluable formalin-fixed paraffin-embedded tumor sections for CDK6 RNA sequencing and further statistical analysis. RNA exome sequencing was performed by Almac with the Illumina TruSeq RNA Sample Preparation kit v2. CDK6 sequencing data were quantile-normalized across samples.
Statistical method and cutoff point selection
The Kaplan–Meier product limit method was used to estimate the survival curve (OS or PFS) for treatment arms in both CDK6 expression low and high subgroup. Unstratified Cox proportional hazard model were performed to compare treatment effect in both CDK6 expression low and high subgroup. HR estimates and 95% CIs from the Cox model were reported on the Kaplan–Meier plot. The cutoff to define CDK6 expression low and high subgroups was derived from an unstratified Cox proportional hazard interaction model by minimizing the treatment by CDK6 interaction P value in terms of OS.
Software
R programming language was used for analysis.
Chemical synthesis
Chemistry general procedures
HPLC spectra for all compounds were acquired using an Agilent 1200 Series system with a diode-array detection detector. Chromatography was performed on a 2.1 × 150 mm Zorbax 300SB-C18 5-μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 ml min−1. The gradient program was as follows: 1% B (0–1 min), 1−99% B (1–4 min) and 99% B (4–8 min). High-resolution mass spectra data were acquired in positive ion mode using an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. NMR spectra were acquired on a Bruker DRX-600 spectrometer (600 MHz 1H, 150 MHz 13C) or a Varian Mercury spectrometer (400 MHz 1H, 100 MHz 13C). Chemical shifts are reported in ppm (δ). Preparative HPLC was performed on Agilent Prep 1200 series with ultraviolet detector set to 254 nm. Samples were injected into a Phenomenex Luna 75 × 30 mm, 5-μm, C18 column at RT. The flow rate was 40 ml min−1. A linear gradient was used with 10% (or 50%) of methanol (A) in H2O (with 0.1 % TFA) (B) to 100% of methanol (A). HPLC was used to establish the purity of target compounds. All final compounds had >95% purity using the HPLC methods described above.
(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)glycine
A solution of 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (1.38 g, 5.0 mmol), tert-butyl glycinate (0.66 g, 5.0 mmol) and N,N-diisopropylethylamine (1.31 ml, 7.5 mmol) in DMF (10 ml) was heated to 85 °C in a microwave reactor for 40 min. After cooling to RT, the reaction was diluted with water and extracted with ethyl acetate (3×). Combined organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by silica gel chromatography to give the desired tBu ester intermediate as oil. This intermediate was treated with a solution of hydrogen chloride in dioxane (10 ml, 4.0 M) for 16 h. The reaction was concentrated under reduced pressure to give the desired acid product (0.24 g, 14%). 1H NMR (600 MHz, CD3OD) δ 7.57 (dd, J = 8.5, 7.1 Hz, 1H), 7.11 (d, J = 7.1 Hz, 1H), 6.95 (d, J = 8.5 Hz, 1H), 5.07 (dd, J = 12.6, 5.5 Hz, 1H), 4.12 (s, 2H), 2.86 (ddd, J = 18.0, 14.4, 5.4 Hz, 1H), 2.74 – 2.67 (m, 2H), 2.15 – 2.08 (m, 1H). MS (ESI) m/z 332.1 [M + H]+.
4-((2-(4-(6-((6-Acetyl-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydropyrido(2,3-d)pyrimidin-2-yl)amino)pyridin-3-yl)piperazin-1-yl)-2-oxoethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (MS140)
To a solution of (2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)glycine (0.017 g, 0.051 mmol) in DCM (10 ml) and DMSO (2 ml) were added palbociclib (0.023 g, 0.051 mmol), 4-methylmorpholine (0.020 g, 0.20 mmol), 1-hydroxy-7-azabenzotriazole (0.0090 g, 0.066 mmol) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (0.013 g, 0.066 mmol). The reaction mixture was allowed to stir at RT for 16 h, before being concentrated under reduced pressure. The resulting residue was purified by reverse-phase prep-HPLC to yield the product (0.028 g, 72%) as a yellow solid. 1H NMR (600 MHz, CD3OD) δ 9.08 (s, 1H), 8.21 (dd, J = 9.6, 2.8 Hz, 1H), 7.86 (d, J = 2.8 Hz, 1H), 7.55 – 7.50 (m, 2H), 7.03 (d, J = 7.0 Hz, 1H), 6.99 (d, J = 8.5 Hz, 1H), 6.04 – 5.97 (m, 1H), 5.08 (dd, J = 12.7, 5.5 Hz, 1H), 4.24 (s, 2H), 3.88 – 3.73 (m, 4H), 3.42 – 3.31 (m, 4H), 2.92 – 2.82 (m, 1H), 2.80 – 2.67 (m, 2H), 2.50 (s, 3H), 2.43 (s, 3H), 2.35 – 2.27 (m, 2H), 2.15 – 2.06 (m, 3H), 1.94 – 1.87 (m, 2H), 1.73 – 1.66 (m, 2H). High-resolution mass spectra (ESI-TOF) m/z: [M + H]+ calculated for C39H41N10O7, 761.3160; found: 761.3150.
Statistics and reproducibility
No data were excluded in our analysis. All data represent single experiments. In animal experiments, the tumors were randomized across treatments, the investigators were not blinded to allocation during experiments and outcome assessment. Statistical analysis was performed using a two-tailed, paired or unpaired Student’s t-tests between two groups. Variation is indicated by mean ± s.d. unless otherwise specified. GraphPad Prism software (v.5) and R v.3.6.3 were used for statistical calculations.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
MS raw files for CDK6 complex analysis are available at the Mass Spectrometry Interactive Virtual Environment (https://massive.ucsd.edu) under ID MSV000086571.
MS files for global protein degradation have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD023137.
Normalized protein quantification data and cell line omics data can be downloaded at DepMap (https://depmap.org/portal/).
Reagents generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application. Source data are provided with this paper.
Code availability
Data analysis was performed in R v.3.6.3 using custom-made or publicly available R packages. The code is available from GitHub (https://github.com/lijin0303/CDK46_expression_dependency). Figure 3e and Extended Data Fig. 3b were generated with R 3.6.3 (Packages taigr, dplyr, tidyr, ggplot2, ggpubr) and corresponding code can be found in https://github.com/lijin0303/CDK46_expression_dependency. Source data are provided with this paper.
References
Sherr, C. J., Beach, D. & Shapiro, G. I. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 6, 353–367 (2016).
O’Leary, B., Finn, R. S. & Turner, N. C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 13, 417–430 (2016).
Otto, T. & Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17, 93–115 (2017).
Finn, R. S. et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 375, 1925–1936 (2016).
Hortobagyi, G. N. et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N. Engl. J. Med. 375, 1738–1748 (2016).
Ingham, M. & Schwartz, G. K. Cell-cycle therapeutics come of age. J. Clin. Oncol. https://doi.org/10.1200/JCO.2016.69.0032 (2017).
Goetz, M. P. et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 35, 3638–3646 (2017).
Im, S. A. et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N. Engl. J. Med. 381, 307–316 (2019).
Knudsen, E. S. & Witkiewicz, A. K. The strange case of CDK4/6 Inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer 3, 39–55 (2017).
Hamilton, E. & Infante, J. R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 45, 129–138 (2016).
Gong, X. et al. Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell 32, 761–776 (2017).
Kim, S. et al. The potent and selective cyclin-dependent kinases 4 and 6 inhibitor ribociclib (LEE011) is a versatile combination partner in preclinical cancer models. Oncotarget 9, 35226–35240 (2018).
Yang, C. et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene https://doi.org/10.1038/onc.2016.379 (2016).
Placke, T. et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 124, 13–23 (2014).
Taipale, M. et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001 (2012).
Vaughan, C. K. et al. Structure of an Hsp90-Cdc37-CDK4 complex. Mol. Cell 23, 697–707 (2006).
Stepanova, L., Leng, X., Parker, S. B. & Harper, J. W. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes CDK4. Genes Dev. 10, 1491–1502 (1996).
Taipale, M. et al. Chaperones as thermodynamic sensors of drug-target interactions reveal kinase inhibitor specificities in living cells. Nat. Biotechnol. 31, 630–637 (2013).
Grbovic, O. M. et al. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl Acad. Sci. USA 103, 57–62 (2006).
Sawai, A. et al. Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res. 68, 589–596 (2008).
Hallett, S. T. et al. Differential regulation of G1 CDK complexes by the Hsp90-Cdc37 chaperone system. Cell Rep. 21, 1386–1398 (2017).
Lai, A. C. & Crews, C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16, 101–114 (2017).
Brand, M. et al. Homolog-selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem. Biol. 26, 300–306 e309 (2019).
Jiang, B. et al. Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angew. Chem. Int. Ed. Engl. 58, 6321–6326 (2019).
Ruscetti, M. et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362, 1416–1422 (2018).
Finn, R. S. et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 11, R77 (2009).
Goldman, J. W. et al. A randomized phase 3 study of abemaciclib versus erlotinib in previously treated patients with stage IV NSCLC with KRAS mutation: JUNIPER. J. Clin. Oncol. 36, 9025 (2018).
Pacheco, J. & Schenk, E. CDK4/6 inhibition alone and in combination for non-small cell lung cancer. Oncotarget 10, 618–619 (2019).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019).
Kato, J. Y., Matsuoka, M., Strom, D. K. & Sherr, C. J. Regulation of cyclin D-dependent kinase 4 (CDK4) by CDK4-activating kinase. Mol. Cell. Biol. 14, 2713–2721 (1994).
McFarland, J. M. et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 9, 4610 (2018).
Meyers, R. M. et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784 (2017).
Tsherniak, A. et al. Defining a cancer dependency map. Cell 170, 564–576 (2017).
McDonald, E. R. 3rd et al. Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 170, 577–592 (2017).
Nusinow, D. P. et al. Quantitative proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387–402 (2020).
Marcotte, R. et al. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell 164, 293–309 (2016).
Gadd, M. S. et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 13, 514–521 (2017).
Chen, P. et al. Spectrum and degree of CDK drug interactions predicts clinical performance. Mol. Cancer Ther. 15, 2273–2281 (2016).
Bondeson, D. P. et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617 (2015).
Winter, G. E. et al. Drug development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015).
Chamberlain, P. P. et al. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 21, 803–809 (2014).
Zhang, C. et al. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK). Eur. J. Med. Chem. 151, 304–314 (2018).
Fischer, E. S. et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014).
Lebraud, H., Wright, D. J., Johnson, C. N. & Heightman, T. D. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent. Sci. 2, 927–934 (2016).
Sumi, N. J., Kuenzi, B. M., Knezevic, C. E., Remsing Rix, L. L. & Rix, U. Chemoproteomics reveals novel protein and lipid kinase targets of clinical CDK4/6 inhibitors in lung cancer. ACS Chem. Biol. 10, 2680–2686 (2015).
Bisi, J. E. et al. Preclinical development of G1T38: a novel, potent and selective inhibitor of cyclin dependent kinases 4/6 for use as an oral antineoplastic in patients with CDK4/6 sensitive tumors. Oncotarget 8, 42343–42358 (2017).
Martinez Molina, D. et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 (2013).
Jafari, R. et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100–2122 (2014).
Patricelli, M. P. et al. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 18, 699–710 (2011).
Verba, K. A. et al. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547 (2016).
Boczek, E. E. et al. Conformational processing of oncogenic v-Src kinase by the molecular chaperone Hsp90. Proc. Natl Acad. Sci. USA 112, E3189–E3198 (2015).
Zhang, J., Yang, P. L. & Gray, N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 (2009).
Bockstaele, L., Bisteau, X., Paternot, S. & Roger, P. P. Differential regulation of cyclin-dependent kinase 4 (CDK4) and CDK6, evidence that CDK4 might not be activated by CDK7, and design of a CDK6 activating mutation. Mol. Cell. Biol. 29, 4188–4200 (2009).
Nomanbhoy, T. K. et al. Chemoproteomic evaluation of target engagement by the cyclin-dependent kinase 4 and 6 inhibitor palbociclib correlates with cancer cell response. Biochemistry 55, 5434–5441 (2016).
Romano, G. et al. A preexisting rare PIK3CA(E545K) subpopulation confers clinical resistance to MEK plus CDK4/6 inhibition in NRAS melanoma and is dependent on S6K1 signaling. Cancer Discov. 8, 556–567 (2018).
Herrera-Abreu, M. T. et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 76, 2301–2313 (2016).
Costa, C. et al. PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kα inhibitors in breast cancer. Cancer Discov. 10, 72–85 (2020).
de Leeuw, R. et al. MAPK reliance via acquired CDK4/6 inhibitor resistance in cancer. Clin. Cancer Res. 24, 4201–4214 (2018).
Li, Z. et al. Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the hippo pathway. Cancer Cell 34, 893–905 (2018).
Guiley, K. Z. et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science https://doi.org/10.1126/science.aaw2106 (2019).
Green, J. L. et al. Direct CDKN2 modulation of CDK4 alters target engagement of CDK4 inhibitor drugs. Mol. Cancer Ther. 18, 771–779 (2019).
Nusinow P. & Gygi, S. P. A guide to the quantitative proteomic profiles of the Cancer Cell Line Encyclopedia. Preprint at bioRxiv https://doi.org/10.1101/2020.02.03.932384 (2020).
Acknowledgements
We thank A. Lujambio, S. Parekh, R. Parsons, E.P. Reddy, R. Sachidanandam, (Icahn School of Medicine at Mount Sinai), I. Aifantis (New York University), S.M. Buckley (University of Nebraska), C.A. Pratilas (Johns Hopkins University) and W.G. Kaelin Jr. (Harvard Medical School) for sharing cell lines and reagents. P.I.P. is supported by the US National Institutes of Health (NIH)/NCI (R01CA204314, R01 CA240362 and R01CA238229), the Irma T. Hirschl Trust, the Manhasset Women’s Coalition against Breast Cancer, the Breast Cancer Alliance, the Melanoma Research Foundation, the Melanoma Research Alliance and Tisch Cancer Institute developmental awards. T.A.A. has been supported by grant T32CA078207 and Z.K. would like to acknowledge the 2017 Robin Chemers Neustein Postdoctoral Fellowship. W.R.S. acknowledges support from the NIH (1R01CA233626) and the Ludwig Center at Harvard, T.I. is supported by the Department of Defense Peer Reviewed Cancer Research Program Horizon Award (W81XWH-19-1-0271). X.C. is supported by the NIH (R01-GM133107). S.A.A. acknowledges support from a grant from the Breast Cancer Research Foundation. This work was supported in part by the P30CA196521 grant (to J.J.) from the NIH and an endowed professorship from the Icahn School of Medicine at Mount Sinai (to J.J.). This work utilized the AVANCE NEO 600 MHz NMR Spectrometer System that was upgraded with funding from an NIH SIG grant 1S10OD025132-01A1.
Author information
Authors and Affiliations
Contributions
X.W. conceived of the study, performed most of the experiments, analyzed the data and helped write the manuscript. X.Y. and Y.X. synthesized chemical compounds. T.A.A, Z.K. and C.A. performed experiments and analyzed data. S.A.A designed research, analyzed data and helped write the manuscript. J.L. and J.J. designed chemical compounds and analyzed data. L.X. carried out MS analysis. B.U. and X.C. supervised MS analysis. R.L., L.W., T.I. and H.W. performed data analysis. S.G.B. and W.R.S. designed the research, analyzed the data and helped to write the manuscript. P.I.P conceived of the study, designed research, analyzed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
X.W., X.Y., Y.X., T.A., Z.K., J.L., J.J. and P.I.P. hold a patent on CDK4/6-directed degradation (WO/2018/106870). The Jian Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics and Cullgen, Inc. J.J. is an equity shareholder and consultant of Cullgen, Inc. H.W. and S.G.B. are employees of Eli Lilly. All other authors declare no competing interests.
Additional information
Peer review information Nature Cancer thanks John Brognard, Keiran Smalley and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Intrinsic resistance to CDK4/6i is associated with incomplete inhibition of Rb/E2F and expression of CDK6.
a, Cell growth crystal violet assay for the indicated cell lines treated with increasing concentrations of PB for 10-16 days and stained with crystal violet. CDK4/6i-sensitive cell lines were highlighted in blue, CDK4/6i-resistant cell lines were in red. b, MCF7 and HCT116 were treated with 1 µM PB for 24, 48 and 72 hr and lysates were immunoblotted with the indicated antibodies. c, Colo205 cells were treated with 1 µM PB at the indicated time points. Cell lysates were immunoblotted with the indicated antibodies.
Extended Data Fig. 2 Low expression of CDK6 predicts for sensitivity to CDK4/6i in NSCLC.
a, The indicated cell lines were treated with increasing concentrations of PB for 24 hr. Lysates were immunoblotted with the indicated antibodies. b, GI90 values of PB and CDK4/6 dependency in NSCLC cell lines.
Extended Data Fig. 3 Tumors expressing both CDK4 and CDK6 depend selectively on CDK6.
a, A673 and TC-71 cells were transfected with non-targeting control or siCDK4 or siCDK6 for 72 hr. Lysates were immunoblotted with the indicated antibodies. b, Relationship between CDK4 and CDK6 expression (CCLE RNA-seq) and DepMap CRISPR–Cas9 single-gene knockout scores (CERES; 20Q1 public dataset). All expression values are in log2(TPM +1). Cell lines harboring COSMIC hotspot mutations to RB1 are annotated in orange. P-values were calculated based on linear regression analysis.
Extended Data Fig. 4 Development of MS140, a potent and selective CDK4/6-degrader (PROTAC).
a. IC50 of in vitro kinase activity assays for PB and MS140 against CDK4/cyclin D1 and CDK6/cyclin D1. b, T47D cells were pretreated with either the proteasome inhibitor 100 nM bortezomib (BOR), 10 µM PB, 10 µM pomalidomide (POM) or 1 µM MLN4924 (MLN) for 4 hr, followed by treatment with MS140 (100 nM/3 hr). Lysates were subjected to immunoblotting with the indicated antibodies. c, Chemical structure of the MS140 negative control (MS140-ve) that does not bind CRBN.
Extended Data Fig. 5 CDK4/6-directed degradation is more effective than CDK4/6i in CDK4/6i-S tumor cells.
a, MCL cell lines were treated with 0.1 µM PB or MS140 at different time points. Lysates were immunoblotted with the indicated antibodies. b, Colo205 cells expressing Dox-inducible shCDK4 or shCDK6 were treated with or without 0.1 µg/ml doxycycline for 72 hr and cell lysates were subjected to immunoblotting with the indicated antibodies. c, Colo205 cells expressing Dox-inducible shCDK4 or shCDK6 were treated with or without 0.1 µg/ml doxycycline for 10 days followed by crystal violet staining. d, Dependency score of CDK4 and CDK6 from cancer cell line encyclopedia (CCLE) and Depmap portal database. e, GI50 values of PB and MS140 in hematologic cancer cell lines. f, Growth curve for an efficacy assay in JeKo-1 tumor xenografts in nude mice treated with vehicle or MS140 (25 mg/kg, b.i.d) or PB (50 mg/kg, q.d.) for 21 days. Each treatment contained 8 animals (n=8). Data represent mean ± SEM. g, Body weight in mice bearing JeKo-1 tumors treated with vehicle (n=8) or PB (50 mg/kg, q.d., n=8) or MS140 (25 mg/kg, b.i.d., n=8) in the course of the experiment (21 days). Data are presented as mean ± S.D. h, White blood cell, lymphocytes and red blood cell counts in C57BL/6 mice before treatment and post treatment with PB (50 mg/kg, q.d., n=8) or MS140 (25 mg/kg, b.i.d., n=7) for 21 days. Data are presented as mean ± S.D. Statistical significance was determined by paired two-tailed Student’s t-test.
Extended Data Fig. 6 In CDK4/6-R cells, CDK4/6 degraders fail to degrade CDK6 due to weak binding of compound.
a, Calu6 cells transiently expressing pcDNA3 (Ev) or pcDNA3-Flag-CRBN were treated with increasing concentrations of MS140 for 24 hr. Lysates were subjected to immunoblotting with the indicated antibodies. b, MV4-11 and A375 were treated MS140 (3 nM) or YKL-06-102 (3 nM) or BSJ-02-162 (3 nM) at different time points. Lysates were subjected to immunoblotting with the indicated antibodies. c, KMS-12-PE and Calu6 were treated MS140 (3 nM) or YKL-06-102 (3 nM) or BSJ-02-162 (3 nM) at different time points. Lysates were subjected to immunoblotting with the indicated antibodies.
Extended Data Fig. 7 CDK4/6i-resistant cells express CDK6 as a thermostable, weak HSP90 client protein.
a, Comparison of total peptide-spectrum match (PSM) for CDK6-interacting proteins by mass spectrometry in KMS-12-PE and Calu6. b, Cell lysates from Colo205 and Calu6 were either subjected to Co-IP with a CDK6 antibody followed by immunoblotting with HSP90, CDC37 and CDK6, or immunoblotted with the indicated antibodies. c, The indicated cell lines were treated with increasing concentrations of Ganetespib (GAN) for 24 hr. Lysates were subjected to immunoblotting with the indicated antibodies. d, The indicated cell lines were treated with 40 nM Luminespib (LUM) at the indicated time points. Lysates were subjected to immunoblotting with the indicated antibodies. e. Calu6 cell line expressing Dox-inducible shCDC37 were treated with or without 0.1 µg/ml doxycycline for 72 hr and cell lysates were subjected to immunoblotting with the indicated antibodies. f, CDK4-dependent cell lines were treated with 30 nM GAN for the indicated time points. Lysates were subjected to immunoblotting with the indicated antibodies. g, KMS-12-PE and Calu6 were treated with increasing concentrations of GAN for 24 hr. Lysates were subjected to immunoblotting with the indicated antibodies. h, Lysates from A375 cells ectopically expressing V5-CDK6 or V5-CDK6 S178p were immunoprecipited with a V5 antibody. The immunoprecipitates were subjected to kinase assay with recombinant Rb protein as substrate.
Supplementary information
Supplementary Table 1
Oligonucleotide sequences
Source data
Source Data Fig. 1
Unprocessed western blots.
Source Data Fig. 2
Unprocessed western blots.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 4
Unprocessed western blots.
Source Data Fig. 5
Unprocessed western blots.
Source Data Fig. 6
Unprocessed western blots.
Source Data Fig. 7
Unprocessed western blots.
Source Data Fig. 1
Statistical Source Data.
Source Data Fig. 2
Statistical Source Data.
Source Data Fig. 3
Statistical Source Data.
Source Data Fig. 4
Statistical Source Data.
Source Data Fig. 5
Statistical Source Data.
Source Data Fig. 6
Statistical Source Data.
Source Data Fig. 7
Statistical Source Data.
Source Data Extended Data Fig. 1
Unprocessed western blots.
Source Data Extended Data Fig. 2
Unprocessed western blots.
Source Data Extended Data Fig. 3
Unprocessed western blots.
Source Data Extended Data Fig. 4
Unprocessed western blots.
Source Data Extended Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 6
Unprocessed western blots.
Source Data Extended Data Fig. 7
Unprocessed western blots.
Source Data Extended Data Fig. 2
Statistical Source Data.
Source Data Extended Data Fig. 3
Statistical Source Data.
Source Data Extended Data Fig. 4
Statistical Source Data.
Source Data Extended Data Fig. 5
Statistical Source Data.
Source Data Extended Data Fig. 7
Statistical Source Data.
Rights and permissions
About this article

Cite this article
Wu, X., Yang, X., Xiong, Y. et al. Distinct CDK6 complexes determine tumor cell response to CDK4/6 inhibitors and degraders. Nat Cancer 2, 429–443 (2021). https://doi.org/10.1038/s43018-021-00174-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-021-00174-z
This article is cited by
-
ERK hyperactivation serves as a unified mechanism of escape in intrinsic and acquired CDK4/6 inhibitor resistance in acral lentiginous melanoma
Oncogene (2024)
-
PROTAC-mediated CDK degradation differentially impacts cancer cell cycles due to heterogeneity in kinase dependencies
British Journal of Cancer (2023)
-
The RNA-binding protein LRPPRC promotes resistance to CDK4/6 inhibition in lung cancer
Nature Communications (2023)
-
LIMK2 promotes melanoma tumor growth and metastasis through G3BP1-ESM1 pathway-mediated apoptosis inhibition
Oncogene (2023)
-
Cellular mechanisms underlying response and resistance to CDK4/6 inhibitors in the treatment of hormone receptor-positive breast cancer
Breast Cancer Research (2022)
