


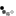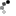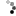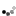
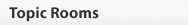







« Prev Next »
Currently, Down syndrome (DS) is one of the most common birth defects, affecting about one in every 750 live births. John Langdon Down first described this condition in the medical literature in 1866, documenting the various symptoms associated with the syndrome but failing to determine their cause. In fact, the cause of DS remained unknown for nearly 100 years following Down's work. Then, in the 1950s, researchers finally determined the source of DS: the presence of an extra copy of chromosome 21, a condition often referred to as trisomy 21.
Since the discovery of trisomy 21, scientists have made great strides in Down syndrome research. For instance, researchers have identified a second (although less common) cause of the condition that is related to chromosomal translocation, and they have also determined the complete DNA sequence of chromosome 21. In addition, scientists have recently created mouse models of DS that possess chromosomal abnormalities similar to those of human patients. Through the use of these models, researchers hope not only to gain a better understanding of the specific genes that play the most significant role in DS, but also to eventually develop improved medical treatments for patients with this condition.
John Langdon Down

Figure 1:
John Langdon Down.
Portrait by Sydney Hodges, ca. 1870, City of London Metropolitan Archives.
During the mid-1800s, English physician John Langdon Down (Figure 1) devoted much of his professional life to children with mental retardation. Early in his career, Down served as medical superintendent at the Royal Earlswood Asylum for Idiots, a charitable establishment dedicated to educating children with learning disabilities. As chief physician, Down noticed that the children at the facility displayed a range of symptoms, and he sought to develop a clinically useful classification scheme to describe these variations. Thus, in his classic paper of 1866, Down noted that many children with mental retardation shared a common set of facial features, including an upward slant to the eye, a flat nose, and a large tongue. He also noted that these children were born with their condition (i.e., the condition was congenital), and that affected patients had a shortened life expectancy. However, Down was unable to determine the cause of this condition, which we now know as Down syndrome, incorrectly attributing it to tuberculosis in an affected child's parents (Down, 1866).
Determining the Causes of Down Syndrome
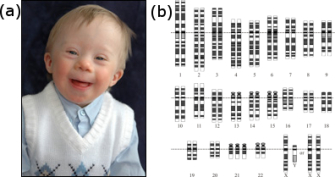
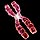
Figure 2: Primary Down syndrome is caused by the presence of three copies of chromosome 21.
(a) A child who has Down syndrome. (b) Idiogram of a person who has primary Down syndrome.
(a) © 2006 iStock.com/JSCook. (b) National Institutes of Health.
As previously mentioned, almost 100 years elapsed between Down's medical description of DS and the discovery of the cause of this condition. But why was this the case? It was certainly not for lack of trying. Many theories for the cause of DS were put forth in the century after Down's publication. Some physicians even made the key observation that older mothers had a higher frequency of DS babies, and they postulated that the condition was caused by what they termed "uterine exhaustion." Gradually, researchers narrowed in on the real cause of DS: a chromosomal abnormality. In fact, researchers now realize that older mothers have more babies with DS because the frequency of meiotic nondisjunction increases in women with age.
The reason that cytologists in the early twentieth century failed to correctly diagnose DS is almost certainly due to technical limitations. Chromosome 21 is the smallest human chromosome, and procedures for examining human chromosomes were still being developed during the first part of the century. Many early cytologists had, in fact, studied chromosomes from DS patients, but none had been able to detect a supernumerary copy of chromosome 21. A breakthrough finally occurred in 1956, when Joe Hin Tjio and Albert Levan described a set of experimental conditions that allowed them to correctly identify the number of human chromosomes as 46. Within three years of the publication of this groundbreaking work, Jerome Lejeune in France and Patricia Jacobs in the United States were able to identify a supernumerary copy of chromosome 21 in karyotypes prepared from DS patients (Figure 2). Trisomy 21 is now accepted to be the major cause of DS, accounting for about 95% of cases.
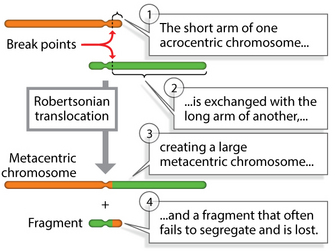
Figure 3: In a Robertsonian translocation, the short arm of one acrocentric chromosome is exchanged with the long arm of another.
© 2014 Nature Education Adapted from Pierce, Benjamin. Genetics: A Conceptual Approach, 2nd ed. All rights reserved. 
Since
the late 1950s, scientists have also determined that a smaller number of DS
cases (roughly 5%) are caused by chromosomal translocations. (Because the translocations responsible for DS
can be inherited, this form of the disease is sometimes referred to as familial
DS.) In these cases, a segment of chromosome 21 is transferred to a second
chromosome, usually chromosome 14 or 15. When the translocation chromosome with
the extra piece of chromosome 21 is inherited together with two normal copies
of chromosome 21, DS results. It is possible, however, for individuals to
inherit a translocation of chromosome 21 without acquiring DS. These
individuals, known as translocation carriers, have inherited both products of
the original translocation event. Hence, translocation carriers have two
chromosomal abnormalities, but the abnormalities balance each other.
Robertsonian translocation, for example, is one of many different types of translocation that cytogeneticists have identified in DS patients over the years. In these cases, Down syndrome is caused by a translocation of chromosomes 15 and 21 (Figure 3), in which the long arms of two acrocentric chromosomes are translocated to produce a single long chromosome, leaving a short one that fails to segregate, thus reducing the overall chromosome number.
The identification of particular translocation defects responsible for DS has been greatly facilitated by the development of banding techniques that allow cytogeneticists to distinguish different breakpoints in chromosomes. From the many karyotypes that cytogeneticists have assembled from DS patients, we now understand that even partial trisomies of chromosome 21 can cause DS. In fact, scientists are interested in determining whether there are critical regions of chromosome 21 specifically responsible for the symptoms of DS.
Mouse Models of Down Syndrome
One could argue that the presence of extra copies of chromosome 21 in DS patients is only a correlation between an abnormality and the disease. However, scientists have developed trisomic mouse models that display symptoms of human DS, providing strong evidence that extra copies of chromosome 21 are, indeed, responsible for DS. It is possible to construct mouse models of DS because mouse chromosomes contain several regions that are syntenic with regions on human chromosome 21. (Syntenic regions are chromosomal regions in two different species that contain the same linear order of genes.) With mapping of the human and mouse genomes now complete, researchers can identify syntenic regions in mouse and human chromosomes with great precision.
As shown in Figure 4, regions on the arms of mouse chromosomes 10 (MMU10), 16 (MMU16), and 17 (MMU17) are syntenic with regions on the long q arm of human chromosome 21. Using some genetic tricks, scientists have induced translocations involving these mouse chromosomes, producing mice that are trisomic for regions suspected to play a role in DS. (Note that these are not perfect models, because the trisomic regions contain many mouse genes in addition to those that are syntenic to human chromosome 21 genes.) These experiments have shown that genes from MMU16 are probably most important in DS, because mice carrying translocations from MMU16 display symptoms more like human DS than mice carrying translocations of MMU10 or MMU17.
Additional experiments have tried to identify particularly important genes within this region by transferring smaller segments of the interval on MMU16. For example, the three mouse models depicted on the right in Figure 4 carry different portions of MMU16, and all display some symptoms of DS. Of the three, the most faithful model of DS is the Ts65Dn mouse, which carries 132 genes that are syntenic with human chromosome 21. This particular mouse demonstrates many of the symptoms of human DS, including altered facial characteristics, memory and learning problems, and age-related changes in the forebrain.
These results are both daunting and promising. On one hand, they suggest that there will be no magic bullet for treating DS, because large numbers of genes are most likely involved in the condition. On the other hand, the results suggest that mouse models will be useful in developing treatments for the many DS patients around the world.
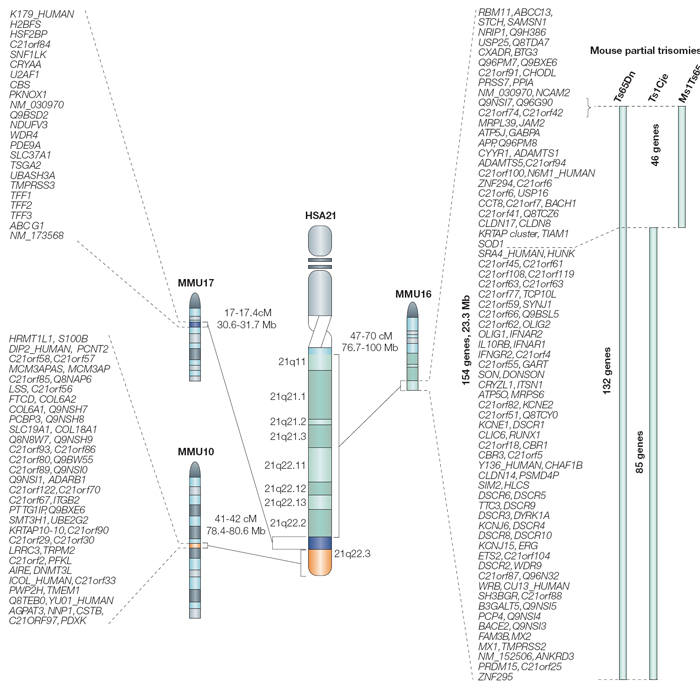
Figure 4: Regions of synteny between human chromosome 21 (HSA21) and mouse chromosomes (MMUs) 16, 17, and 10.
There are three partial trisomy mouse models of human trisomy 21, all trisomic for a portion of MMU 16. The gene content of these partial trisomies is shown on the right.
© 2004 Nature Publishing Group Antonarakis, S. E. et al. Chromosome 21 and Down syndrome: from genomics to pathophysiology. Nature Reviews Genetics 5, 730 (2004). All rights reserved.
Summary
Despite the questions raised by ongoing genetic studies of DS, one thing remains certain: Researchers have come a long way in their understanding of this condition since John Langdon Down first described its symptoms in 1866. For instance, scientists now know that the vast majority of DS cases involve trisomy 21, although approximately 5% arise as a result of various chromosomal translocations. Moreover, thanks to areas of synteny between the human and mouse genomes, researchers have also been able to use mouse models to pinpoint the specific genes on human chromosome 21 that are most likely involved in producing symptoms of DS. In the future, scientists will no doubt continue to focus on these genes with the ultimate goal of creating new, DNA-based treatments for Down syndrome. Furthermore, they will likely expand these methods to the study of other trisomies beyond trisomy 21.
References and Recommended Reading
Antonarakis, S. E., et al. Chromosome 21 and Down syndrome: From genomics to pathophysiology. Nature Reviews Genetics 5, 725–738 (2004) doi:10.1038/nrg1448 (link to article)
Down, J. L. Observations on an ethnic classification of idiots. London Hospital Reports 3, 259–262 (1866) (link to article)
Hattori, M., et al. The DNA sequence of human chromosome 21. Nature 405, 311–319 (2000) doi:10.1038/35012518 (link to article)
Patterson, D., & Costa, A. C. S. Down syndrome and genetics—A case of linked histories. Nature Reviews Genetics 6, 137–147 (2005) doi:10.1038/nrg1525 (link to article)