
Abstract
Sordarin is a glycoside antibiotic with a unique tetracyclic diterpene aglycone structure called sordaricin. To understand its intriguing biosynthetic pathway that may include a Diels-Alder-type [4+2]cycloaddition, genome mining of the gene cluster from the draft genome sequence of the producer strain, Sordaria araneosa Cain ATCC 36386, was carried out. A contiguous 67 kb gene cluster consisting of 20 open reading frames encoding a putative diterpene cyclase, a glycosyltransferase, a type I polyketide synthase, and six cytochrome P450 monooxygenases were identified. In vitro enzymatic analysis of the putative diterpene cyclase SdnA showed that it catalyzes the transformation of geranylgeranyl diphosphate to cycloaraneosene, a known biosynthetic intermediate of sordarin. Furthermore, a putative glycosyltransferase SdnJ was found to catalyze the glycosylation of sordaricin in the presence of GDP-6-deoxy-d-altrose to give 4′-O-demethylsordarin. These results suggest that the identified sdn gene cluster is responsible for the biosynthesis of sordarin. Based on the isolated potential biosynthetic intermediates and bioinformatics analysis, a plausible biosynthetic pathway for sordarin is proposed.
Similar content being viewed by others
Introduction
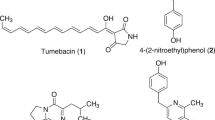
Sordarin 1 (Figure 1) is a diterpene glycoside produced by the mold Sordaria araneosa Cain1 and is known as a unique antifungal agent targeting elongation factor 2 in eukaryotes (eEF2).2, 3 Elongation factor 2 in eukaryotes catalyzes the translocation of transfer RNA and messenger RNA after peptide bond formation in the translation process and is a unique target to combat fungal infections. Thus, extensive structural activity relationship studies of sordarin have been undertaken to identify compounds with improved antifungal activity4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 and to investigate its mode of action.20 In addition, its unique tetracyclic diterpene structure and biological activity have generated interest in the development of synthetic methodology useful for its total synthesis.21, 22, 23, 24

Isolated sordarin related metabolites from S. araneosa and the hypothesized biosynthetic pathway. The 14C-labeled cycloaraneosene 2 was incorporated into sordarin 1. Compounds 1–6 except for 3 were isolated from the sordarin producer Sordaria araneosa in the present study. Cycloaraneosene derivative 6 was identified for the first time from the culture of this strain.
From a biosynthetic study of sordarin, Borschberg showed that [2-14C]mevalonate is incorporated into the core structure of 1 (Figure 1).25 In addition, [18-14C]cycloaraneosene was incorporated into 1 indicating that cycloaraneosene 2 is a biosynthetic intermediate for 1 (Figure 1).25 These incorporation studies suggest that 2 is constructed from geranylgeranyl diphosphate (GGDP) via the mevalonate pathway and certain cycloaraneosene derivatives are converted into the unique sordaricin skeleton (Figure 1). Cycloaraneosene 2 has a characteristic 5-8-5 tricyclic diterpene structure that is similar to, but distinct from fusicocca-2,10(14)-diene in fusicoccin A biosynthesis26 and cyclooct-9-en-7-ol in cyclooctatin biosynthesis (Supplementary Figure S1).27 Diterpene cyclases specific for fusicocca-2,10(14)-diene (PaFS) and cyclooct-9-en-7-ol (CotB2) have been already characterized.26, 27 In addition, a detailed mechanistic study of the CotB2 reaction has been recently reported.28, 29 The distinct cyclization mechanism of these unique diterpene cyclases is intriguing from a mechanistic standpoint.
Transformation of the cycloaraneosene derivative to sordaricin 3 has been proposed to include a Diels-Alder type [4+2]cycloaddition (Figure 1).30, 31 In the proposed biosynthetic reaction mechanism, the C8–C9 bond of the cycloaraneosene skeleton is cleaved via a Baeyer–Villiger-type oxidation to give a carboxylate and an aldehyde as functional groups. The generated acrolein moiety acts as a dienophile and reacts with the cyclopentadiene moiety via a [4+2]cycloaddition to give 3. The Mander and Kato groups demonstrated that this cycloaddition can occur with synthetic analogs, but usually harsh conditions and a long reaction time are required.31, 32, 33, 34 Thus, an enzyme mediated [4+2]cycloaddition mechanism seems to be involved in the formation of 3 from the cycloaraneosene derivative. In the present study, to understand the unprecedented biosynthetic reaction mechanisms involved in the biosynthesis of 1, a genome-mining approach was used to identify the corresponding biosynthetic genes.
Results
Identification of sordarin related metabolites in Sordaria araneosa
To ensure the production of 1 by S. araneosa Cain ATCC 36386, its metabolites including sordarin 1 were isolated and the structures were determined (Figure 1, Supplementary Figures S2–S15). Hypoxysordarin 4,35 which is an acylated derivative of sordarin, was found to be a major metabolite in our culture conditions and appears to be a dead-end metabolite in the biosynthesis (Supplementary Figures S4 and S5). Three cycloaraneosene derivatives, cycloaraneosene 2 (Supplementary Figures S6 and S7),25, 36 cycloaraneosen-8-ol 5 (Supplementary Figures S8 and S9),25, 37 and cycloaraneosene-8,9,13-triol 6 (Supplementary Figures S10–S15,Supplementary Table S1) were also isolated. In addition to 2, 5 and 6 seemed to be biosynthetic intermediates of 1. The stereochemistry of 6 was determined with nuclear Overhauser effect (NOE) analysis based on the stereochemistry of 5 (Supplementary Figure S15). The target biosynthetic gene cluster for sordarin/hypoxysordarin was thus expected to encode a diterpene cyclase, a glycosyltransferase, several hydroxylases such as cytochrome P450 monooxygenase, and a polyketide synthase (PKS) because a considerable amount of the polyketide hybrid-type compound, hypoxysordarin 4, was isolated.
Genome mining of the sordarin biosynthetic gene cluster
Draft genome sequence analysis of the genomic DNA of S. araneosa was performed with the Illumina Genome Analyzer (GA) IIx. The obtained read sequences were assembled to determine the total 45 Mb of the DNA sequences in 103 scaffolds. The size of the genome sequence is similar to that of Neurospora crassa (ca. 40 Mb)38 and Sordaria macrospora (ca. 40 Mb).39 Thus, the determined draft sequence seems to cover the full length of the genome sequence of S. araneosa. In silico screening of the draft genome sequence with PaFS, which is a bi-functional enzyme involved in fusicoccin biosynthesis with a N-terminus diterpene cyclase domain and C-terminus prenyltransferase domain,26 indicated that two regions in the genome of S. araneosa might encode a similar prenyltransferase (data not shown). The coding sequences adjacent to the two regions were then carefully predicted using Augustus software with Neurospora crassa as the model organism.40 One of the regions was found to encode a putative terpene cyclase, a type I PKS, a glycosyltransferase and six cytochrome P450 monooxygenases (Table 1). No other region with this feature could be found in the obtained draft genome sequence. We then named this region (ca. 67 Kb) as the sordarin biosynthetic gene (sdn) cluster.
Functional characterization of the diterpene cyclase SdnA
To support that the identified sdn gene cluster is responsible for the sordarin/hypoxysordarin biosynthesis, in vitro functional analysis of two characteristic enzymes, a putative terpene cyclase SdnA and a putative glycosyltransferase SdnJ, were conducted. Total RNA was extracted from the harvested cells and the complementary DNA (cDNA) library was constructed as a template for PCR. The sdnA and sdnJ genes were amplified by PCR with the cDNA library and successfully expressed in E. coli (Supplementary Figure S16). The purified SdnA was reacted with GGDP and incubated at 30 °C for 16 h according to the related diterpene cyclase reaction conditions.41 The reaction product was extracted with hexane and the extract was analyzed by gas chromatography–mass spectrometry. A signal was detected at 12.6 min with a m/z 272, which corresponds to the molecular ion [M+] of 2 (Figure 2). In the absence of SdnA or MgCl2, this reaction did not occur, indicating that SdnA requires Mg2+ for the reaction. This observation agrees with the general properties of terpene cyclases.42 Five 150-ml-scale enzyme reactions gave 5.7 mg of the product whose structure was unambiguously confirmed as 2 by NMR spectroscopy (Supplementary Figures S6 and S7).25, 36 The optical rotation of the SdnA reaction product from GGDP was [α]D16 −22 (c 0.22, CHCl3), which is similar to the reported data of [α]D −38.4 (c 2.8, CHCl3),25, 36 indicating that the stereochemistry of the SdnA reaction product is the same as the reported cycloaraneosene structure. Thus, SdnA was confirmed to be a cycloaraneosene synthase and seemed to be responsible for sordarin biosynthesis in the producer strain.

Functional analysis of cycloaraneosene synthase SdnA by gas chromatography–mass spectrometry. (a) Authentic cycloaraneosene (natural product), (b) extract of the SdnA reaction with 18 μM of GGDP, 0.8 μM of SdnA and 5 mM of MgCl2 at 30 °C for 16 h, (c) without SdnA. Off set is the mass for the signal at 12.6 min.
Functional characterization of the glycosyltransferase SdnJ
A putative glycosyltransferase SdnJ was speculated to catalyze the glycosylation of 3 in the presence of a nucleotide-diphospho (NDP)-sugar, presumably NDP-6-deoxy-d-altrose, from the chemical structure of 1. The O-methyl group on the 4′-hydroxy group of the sugar moiety (sordarose) in sordarin seems to be introduced after the glycosylation, as this reaction order was reported in the biosynthesis of other O-methylated glycosides such as spinosyn,43 tylosin,44 mycinamycin,45 elloramycin,46 erythromycin,47 oleandomycin48 and FD-594.49 To examine this hypothetical glycosylation by SdnJ, NDP-6-deoxy-d-altrose was chemically synthesized (Supplementary Figure S17). Compound 3 was also prepared from 1 by acid hydrolysis.1, 19 Because SdnI shows high similarity to GDP-mannose-4,6-dehydratase,50, 51, 52 GDP seems to be the most likely nucleotide for this particular deoxysugar biosynthesis.
The recombinant SdnJ protein was successfully expressed in E. coli and purified for the enzymatic reaction analysis (Supplementary Figure S16). SdnJ was mixed with GDP-6-deoxy-d-altrose 7 and 3 at 28 °C for 48 h. After the reaction, the enzymatic reaction product was extracted with ethyl acetate and analyzed by liquid chromatography–electrospray ionization mass spectrometry (Figure 3). A new signal with m/z 501, which corresponds to [M+Na]+ for the expected glycosylated product, was observed only in the presence of all components. This result suggests that SdnJ catalyzes the glycosylation of 3 with 7 to give 4′-O-demethylsordarin 8 (Figure 4). A small amount of glycosylated compound was produced in the presence of ADP-6-deoxy-d-altrose, but no product was detected with TDP-6-deoxy-d-altrose (Figure 3). GDP-d-mannose was also accepted as the glycosyl donor to give mannosyl sordaricin (m/z 517, [M+Na]+), but GDP-d-glucose was not accepted (Supplementary Figure S18). Thus, we concluded that, (1) the guanine moiety of the NDP-sugar as the glycosyl donor is important for recognition, (2) the stereochemistry at C-2 of the GDP-sugar is critical and (3) the existence of a hydroxy group at C-6 and the stereochemistry at C-3 of the GDP-sugar are not important. The characterized enzymatic function of SdnJ indicates that SdnJ is responsible for the glycosylation of 3 with 7 to give 8 in sordarin biosynthesis (Figure 4). Overall, the characterized functions of the cycloaraneosene synthase SdnA and the sordaricin:GDP-6-deoxy-d-altrose glycosyltransferase SdnJ supports that the sdn gene cluster identified by genome mining is responsible for the biosynthesis of sordarin/hypoxysordarin.

Functional analysis of the GDP-6-deoxyaltrose transferase SdnJ by liquid chromatography–electrospray ionization–mass spectrometry (LC–ESI–MS) analysis. The reaction was carried out with (a) GDP-6-deoxy-d-altrose, (b) ADP-6-deoxy-d-altrose and (c) TDP-6-deoxy-d-altrose. The upper panel is the traces for the selected ion m/z 355 for [M+H]+ of sordaricin 3 and the bottom panel is the traces for the selected ion m/z 501 for [M+H]+ of 4′-O-demethylsordarin 8.

The sordarin biosynthetic gene (sdn) cluster and the proposed biosynthetic pathway for sordarin/hypoxysordarin. (a) The biosynthetic pathway for sordarin/hypoxysordarin, (b) GDP-6-deoxyaltrose biosynthesis and (c) polyketide biosynthesis. ACP, acyl carrier protein; AT, acyltransferase; CAT, choline/carnitine acyltransferase; DH, dehydratase; ER, enoylreductase; KR, β-ketoreductase; KS, β-ketoacylsynthase. The ER domain of SdnO should be inactive based on the chemical structure of hypoxysordarin 4.
Discussion
A genome-mining approach is widely used to identify target natural product biosynthetic gene clusters.53, 54, 55 In the present study, we succeeded in identifying the biosynthetic gene cluster responsible for the biosynthesis of the diterpene glycoside antibiotic sordarin/hypoxysordarin from the draft genome sequence of the producer mold Sordaria aragonesa. Bioinformatic analysis of other fungal genome sequences showed that a grapevine pathogen Eutypa lata56 and the fungus Trichoderma harzianum57, which possesses beneficial bio-control activity against fungal plant pathogens, have almost the same contiguous gene cluster from sdnA to sdnO in their genomes. Thus, these fungi might have the ability to produce sordarin/hypoxysordarin-related diterpene glycosides, although these types of compounds have never been isolated from these strains. Three Xylariale families, Xylariaceae, Amphisphaeriaceae and Diatrypaceae, were found to produce sordarin analogs and therefore may contain similar biosynthetic gene clusters in their genomes.58
Based on the annotated functions of the sdn genes as determined by BLAST, a plausible biosynthetic pathway is proposed in Figure 4. First, a putative GGDP synthase, SdnC, constructs GGDP from farnesyl diphosphate and isopentenyl diphosphate. A diterpene cyclase, SdnA, catalyzes the cyclization of GGDP to afford 2, which was verified in the present report. Compound 2 is then hydroxylated four times by the putative cytochrome P450 monooxygenases SdnB, SdnE, SdnF and SdnH to give a hydroxylated cycloaraneosene derivative such as cycloaraneosene-8,9,13,19-tetraol. Although the order of the hydroxylations is unclear, the existence of 5 and 6 in the culture of S. aragonesa suggests that at least C8, C9 and C13 of the cycloaraneosene skeleton are hydroxylated before the sordaricin formation. Dehydration of the 13-hydroxy group of the hydroxylated cycloaraneosene derivative might be catalyzed by an unassigned hypothetical protein such as SdnG and SdnP to construct the cyclopentadiene moiety. The putative flavin adenine dinucleotide-dependent oxidoreductase SdnN is proposed to catalyze the oxidation at C9 of the hydroxylated cycloaraneosene derivative and also catalyze the Baeyer–Villiger oxidation to give the lactone intermediate. Baeyer–Villiger monooxygenases with a flavin cofactor are known to be involved in pentalenolactone biosynthesis59, 60 and the steroid degradation process.61, 62 The presumed lactone intermediate would be hydrolyzed to give an acrolein moiety and a carboxylate moiety. Then, [4+2]cycloaddition would occur between the acrolein moiety and the cyclopentadiene moiety to give 3. SdnN might also be involved in the [4+2]cycloaddition after the hypothesized oxidation to accommodate the oxidized product and prompt the [4+2]cycloaddition. It is known that the flavin-dependent oxidase Sol5 catalyzes the oxidation of an alcohol of prosolanapyrone II and also is involved in the cycloaddition to give solanapyrone A in solanapyrone biosynthesis.63 The oxidation of the alcohol to an aldehyde changes the lowest unoccupied molecular orbital level of the dienophile and the [4+2]cycloaddition seems to occur simultaneously at the active site of Sol5. Although several types of enzymatic [4+2]cycloadditions have been reported,64, 65 the hypothesized mechanism of SdnN has not been recognized.
GDP-6-deoxy-d-altorose 7 may be biosynthesized from GDP-d-mannose by the putative GDP-mannose-4,6-dehydratase SdnI and the short-chain dehydrogenase/reductase SdnK, which is related to GDP-l-fucose biosynthesis (Figure 4b).66, 67 GDP-l-fucose synthase catalyzes both the 3,5-epimerization of GDP-4-keto-6-deoxymannose and reduction at C-4.68, 69 Thus, it would be interesting to know whether the putative reductase SdnK could catalyze both epimerization at C-3 of GDP-4-keto-6-deoxymannose and the subsequent reduction to give 7. The glycosyltransferase SdnJ catalyzes the attachment of 6-deoxy-d-altorose onto the 19-hydroxy group of 3 to give 4′-O-demethylsordarin 8, which was produced by biotransformation of 1 with Streptomyces capreolus and Streptomyces avermitilis.70 A putative methyltransferase SdnD would complete the biosynthesis of 1.
The unique acyl chain at the 3′-hydroxy group of 4 would be constructed by an iterative type I PKS SdnO and the trans-acting polyketide methyltransferase (MT) SdnL (Figure 4c). A trans-acting C-MT of elongated polyketide chain in pseurotin biosynthesis has been recently characterized.71 Thus, SdnL would be responsible for the introduction of an α-methyl group of the polyketide chain. SdnO consists of a β-ketosynthase domain, acyltransferase domain, dehydratase domain, unassigned region, enoylreductase domain, β-ketoreductase domain, acyl carrier protein (ACP) and a choline/carnitine acyltransferase domain. Although intensive studies on fungal and bacterial PKSs have been performed,72 it is still hard to predict the precise polyketide product from only the sequence of SdnO. It is presumed that iterative reactions involving the PKS SdnO and C-MT SdnL would construct certain triene tetraketides whose stereochemistry might be a Z-E-Z configuration judging from the stereochemistry of 4. Thus, SdnO would not catalyze the enoyl reduction similar to the inactive enoylreductase domain of LovB in the PKS of lovastatin biosynthesis.73 The polyketide chain might be transferred to the 3′-hydroxy group of 1 by the putative choline/carnitine acyltransferase domain of SdnO because there is no obvious acyltransferase of the polyketide chain in the sdn cluster. If this is true, it would be a novel completion mechanism of a PKS.74 Alternatively, the putative β-lactamase protein SdnR might be responsible for the cleavage and transfer of the polyketide chain from the PKS SdnO to 1, as a related completion mechanism of a PKS reaction has been reported.75 Two putative cytochrome P450s, SdnQ and SdnT, might catalyze the epoxidations of the polyketide chain to complete the biosynthesis of 4. Transcriptional regulators SdnM and SdnS are presumably encoded for the transcriptional regulation of the expression of the sdn gene cluster.
Cycloaraneosene synthase SdnA shows less similarity to the N-terminus diterpene cyclase domain of PaFS in fusicoccin biosynthesis and rather higher similarity to sesquiterpene synthases such as koraiol synthase (FFUJ_12585, 38% identity/57% similarity).76 This suggests that the functional prediction of terpene cyclases from only the protein sequence is still difficult for annotation. In fact, only the C-terminus GGDP synthase domain of PaFS was useful as a query to fish out the presently identified sdn gene cluster. The reaction mechanism of cycloaraneosene synthase SdnA may be related to that of CotB2, which constructs cycloocta-9-en-7-ol from GGDP (Supplementary Figure S1).28 Because there is little sequence similarity between SdnA and CotB2, detailed mechanistic analysis of SdnA is needed to understand the reaction mechanism.
Characterization of SdnJ as a GDP-6-deoxy-d-altrose transferase of 3 further supports that the identified gene cluster is responsible for sordarin biosynthesis. Addition of ethylenediaminetetraacetic acid (EDTA) did not inhibit the glycosylation between 7 and 3 indicating that SdnJ does not require divalent metal cations for catalysis. Because SdnJ sequentially belongs to the family of GT-B, its lack of requirement for a metal agrees well with the biochemical properties of this family of glycosyltransferases.77 The optimal pH was around pH 7.5 and the optimal temperature was 30 °C. SdnJ recognizes both 7 and GDP-d-mannose as glycosyl donors, although the majority of isolated natural products from S. araneosa contain only 6-deoxy-d-altrose as the sugar part. Thus, 7 would be efficiently biosynthesized and selectively transferred to 3 by SdnJ under physiological conditions. Because 8 shows a higher minimal inhibitory concentration (MIC) value than 1,70 the existence of the 4′-O-methyl group of 1 appears to be remarkable for bioactivity and thus, the O-methylation of the 4′-hydroxy group of the 6-deoxy-d-altlose moiety would be an important biosynthetic step to produce the bioactive molecule.
In conclusion, the sordarin/hypoxysordarin biosynthetic gene cluster was successfully identified by a genome-mining approach. Further, two characteristic enzymes, cycloaraneosene synthase SdnA and GDP-6-deoxy-d-altrose:sordaricin glycosyltransferase SdnJ, were functionally characterized and the results were used to support that the sdn gene cluster is responsible for the biosynthesis of sordarin/hypoxysordarin. Although the transformation mechanism from the cycloaraneosene derivative to the sordaricin skeleton is still unclear, further investigation of the sdn genes using in vitro and in vivo analyses should provide more information in due course.
Materials and Methods
Isolation of metabolites from S. araneosa Cain ATCC 36386
Sordaria araneosa Cain ATCC 36386 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). S. araneosa was inoculated on agar plates of ATCC Medium #350 Emerson YpSs agar (yeast extract 0.4%, soluble starch 1.5%, K2HPO4 0.1%, MgSO4·7H2O 0.05%, agar 20% in 200 ml of tap water (pH 7.0), and autoclaved) and incubated at 26 °C for 7 days. The filamentous fungus was scraped from the agar plate and suspended in 20% glycerol and stored at −80 °C until use. An aliquot of the suspension was inoculated on a new Emerson YpSs agar plate and incubated at 26 °C for 7 days. A 5-mm cube of the agar culture of S. araneosa was then inoculated into a 500-ml-baffled Erlenmeyer flask of a pre-culture medium (potato (freshly grated) 10%, sucrose 2.0%, casamino acid 1.0%, K2HPO4 0.5% in 80 ml of tap water and autoclaved) with a cotton plug and cultured at 26 °C, 200 r.p.m., for 5 days. A portion (ca. 5 ml) of the pre-cultured fungus was transferred into 12 500-ml-baffled Erlenmeyer flasks containing 100 ml of production medium (glucose 10%, polypeptone 1.5%, corn steep liquor 1.0%, yeast extract 0.5%, l-tryptophan 0.2%, K2HPO4 0.5%, FeSO4·7H2O 0.4%, CoSO4 0.05%, MgSO4·7H2O 0.1% in tap water and autoclaved) with a cotton plug and cultured at 26 °C, 200 r.p.m., for 10 days. The cultured cells (158 g from a 1.2 l of culture) were collected by centrifugation (6000 g × 30 min) and then disrupted with acetone (1 l). The acetone-cell suspension was filtered and the solvent was removed on a rotary evaporator. The residue was extracted with ethyl acetate (300 ml × 3 times) and water (300 ml). The combined organic layers were dried over anhydrous MgSO4. The organic solution was concentrated on a rotary evaporator. The residue was purified by silica-gel chromatography with hexane-EtOAc as the eluent. The fractions containing 1, hypoxysordarin 4 and cycloaraneosene-8,9,13-triol 6 were further purified by ODS (Wakogel 50C18) chromatography with CH3OH-H2O as the eluent. A fraction containing 2 and cycloaraneosen-8-ol 5 was further purified by silica-gel chromatography with hexane-EtOAc and CHCl3-hexane as the eluents. From the 1.2 l of culture, 30 mg of 1, 171 mg of 4, 8.4 mg of 6, 5.6 mg of 2 and 3.5 mg of 5 were obtained. See Supplementary Information for their nuclear magnetic resonance spectra (Supplementary Figure S2–S15).
Genome mining of sordarin biosynthetic genes
S. araneosa was grown in a modified yeast extract-malt extract (YEME) medium (yeast extract 0.3%, peptone 0.5%, malt extract 0.3%, glucose 1% and sucrose 34%, after autoclaving 0.5% glycine and 5 mM MgCl2 were added (final concentrations)) at 26 °C, 200 r.p.m., for 6 days. The cultured cells were collected by centrifugation (6000 g × 30 min) and washed with a 10.3% sucrose solution. The resultant cells (8.6 g from 100 ml of culture) were suspended in 1 × TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0) and then treated with 50 mg of zymolyase 20 T (Seikagaku Corporation, Tokyo, Japan) at 45 °C. After gentle incubation for 1 h, 2.7 ml of 0.5 M EDTA, 2.7 ml of H2O and 1.9 ml of 10% sodium dodecyl sulfate solution were added and the solution was incubated for 1 h at 37 °C. The solution was then gently mixed with 1 volume of phenol/CHCl3. After centrifugation, the water layer was extracted and further washed with CHCl3. The obtained water solution was treated with 30 μl RNase for 0.5 h at 37 °C. After the phenol-CHCl3 and CHCl3 extraction, the water layer was mixed with 0.1 volume of 3 m NaOAc and 1 volume of 2-propanol. After gentle mixing, the genomic DNA was recovered with a glass rod. The genome DNA was then dissolved in 10 ml of Tris-NaCl-EDTA (TNE) buffer (1 × TE, 0.1 M NaCl) and then 0.1 volume of 3 M NaOAc and 2.2 volume of EtOH were added. The genomic DNA was recovered with a glass rod and dissolved in 0.5 ml of TE buffer (1.4 mg ml−1) and stored at 4 °C. Draft genome sequence analysis was performed using the Illumina Genome Analyzer (GA) IIx (San Diego, CA, USA) after treatment of the extracted genomic DNA with NEBNext DNA Sample Prep Reagent Set 1 or Mate Pair Library Prep Kit v2 from Takara Bio (Ohtsu, Japan). The obtained read sequences were assembled with Edena, Velvet or ALLPATHS-LG to determine total 45 Mb of the draft DNA sequences in 103 scaffolds. In silico screening of the draft genome sequence with PaFS26 was performed using Geneious software (Biomatters, Auckland, New Zealand) and BioEdit. The coding sequences were predicted by Augustus software40 with Neurospora crassa as a model organism. After cloning of the sdnA and sdnJ genes, coding sequences were revised using Augustus software. A part of the genome DNA sequence containing the sordarin biosynthetic gene (sdn) cluster (ca. 120 kb) was deposited in the DDBJ under the accession number LC079035.
Characterization of diterpene cyclase SdnA
Total RNA was extracted from the harvested cells cultured in the above-mentioned modified YEME medium after 96 h. The cells were suspended in 1 × TE buffer and treated with zymolyase 20 T at 45 °C for 1 h. After centrifugation, the spheroplasts were extracted with Nucleospin RNA II kit (Takara) according to the manufacture’s protocol. Using the obtained total RNA, a cDNA library was constructed using the PrimeScript II 1st strand cDNA Synthesis kit (Takara). The sdnA gene was amplified by PCR with the cDNA library as the template and successfully expressed in E. coli. For the sdnA gene, sdnA-N: 5′-CGCATATGTCACTATACGGGTTATTC-3′ and sdnA-C: 5′-CTACAAGCTTTCCCAAAGGACCG-3′ were used as the PCR primers and were synthesized by Fasmac (Kanagawa, Japan). PrimeSTAR GXL DNA polymerase (Takara) was used for PCR and the reaction solution contained 5 × PrimeSTAR GXL buffer 2 μl, 2.5 mM dNTP mixture 0.8 μl, 100 μM of primers 1.5 μl each, cDNA solution 1.0 μl, sterilized water 3.0 and 0.2 μl of polymerase. The PCR conditions were 10 s of denaturation at 98 °C, 15 s of annealing at 60 °C and 70 s of extension at 68 °C for 40 cycles. After agarose gel electrophoresis, the amplified DNAs were recovered using the FastGene Gel/PCR extraction kit (Nippon Genetics, Tokyo, Japan). The obtained DNA fragments (8.8 μl each) were reacted with Ex Taq polymerase (Takara, 0.1 μl), dNTP mixture (0.2 μl) and EX Taq buffer (1.0 μl) at 70 °C for 2 min. The A attached DNA fragments were then ligated with T-vector pMD19 (Takara) using the DNA ligation kit version 2.1 (Takara) and introduced into E. coli DH5α competent cells. The desired plasmid containing the matured gene (sdnA/pMD19) was confirmed by sequence analysis (Fasmac). The plasmid sdnA/pMD19 was digested with NdeI and HindIII and the insert DNA was introduced into the corresponding site of the pColdI expression vector and transformed into E. coli DH5α. The obtained expression plasmid sdnA/pColdI was then introduced into E. coli BL21(DE3).
The resulting sdnA/pColdI/BL21(DE3) strain was cultured in Luria Bertani medium containing 50 μg ml−1 of ampicillin at 37 °C and 200 r.p.m. This was done until the OD600 reached 0.6 and then isopropyl 1-thio-β-d-galactopyranoside (final concentration 0.2 mM) was added and culture continued at 15 °C overnight. The E. coli cells were harvested by centrifugation at 4700 g for 20 min, and then suspended in 50 mM Tris buffer (pH 7.5, buffer A). The cell suspension was treated with a Q55 sonicator (Qsonica, WAKENBTECH, Kyoto, Japan) to obtain the cell-free extract. Following centrifugation at 13 200 g for 20 min, the supernatant was loaded onto a TALON resin (Takara) column and washed with buffer A containing 20 mM imidazole. The SdnA protein was eluted with buffer A containing 200 mM imidazole. The solution was passed through a PD-10 column (GE Healthcare, Buckinghamshire, UK) to obtain purified SdnA (80 μg from 200 ml of culture). To confirm the size of the recombinant SdnA (predicted size 42 kDa), sodium dodecyl sulfate–polyacrylamide gel electrophoresis (12.5%) was performed. The protein concentration (A280 1 A/cm=1 mg ml−1) was estimated using a Nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
The SdnA enzyme reaction solution consisted of SdnA (0.8 μM), geranylgeranyl diphosphate (GGDP, 17.8 μM), MgCl2 (5 mM), EDTA (0.5 mM) and DTT (2 mM) in Tris buffer (50 mM, pH 7.5, 1 ml). GGDP was prepared by chemical synthesis from geranylgeraniol.78 The reaction was conducted at 30 °C for 16 h and then extracted with hexane three times. After concentration, the extract was analyzed using a gas chromatography–mass spectrometry (GCMS-QP2010, Shimadzu, Kyoto, Japan) equipped with a DB-1 column (0.25 mm × 30 m, Agilent Technologies, Santa Clara, CA, USA). The temperature program for the GC was 60 °C for 2 min, 60–150 °C at 30 °C min−1, 150–180 at 10 °C min−1 and 180–210 at 2 °C min−1. To obtain a larger amount of product, the 150-ml-scale enzymatic reaction with SdnA (17.6 μM, 11 mg), GGDP (296 μM, 20 mg), MgCl2 (1 mM), EDTA (0.5 mM) and DTT (2 mM) in Tris buffer containing 0.1% Triton X-100 (50 mM, pH 7.5) was carried out five times at 30 °C for 24 h. After extraction with hexane and concentration, the extract was purified by silica-gel chromatography with hexane as the solvent. A total of 5.7 mg of cycloaraneosene 2 was obtained. The optical rotation of the SdnA reaction product was analyzed using a JASCO DIP-360 polarimeter (JASCO, Tokyo, Japan).
Preparation of GDP-6-deoxy-d-altrose
The synthetic procedure is described in the Supplementary Information.
Characterization of glycosyltransferase SdnJ
The expression conditions for the sdnJ gene were same as for the sdnA gene, except for the PCR primer, sdnJ-N: 5′-CCATATGCACGCCAAACGCC-3′ and sdnJ-C: 5′-AGTGGATCCATTTGAGGAAACATCG-3′. The E. coli cells containing the recombinant SdnJ protein were suspended in 50 mM Tris buffer with 10% glycerol (pH 7.5, buffer B) and the cell suspension was sonicated to obtain the cell-free extract. Following centrifugation at 13 200 g for 20 min, the supernatant was loaded onto a TALON resin column and washed with buffer B containing 20 mM imidazole. The SdnJ protein was eluted with buffer B containing 200 mM imidazole. The solution was concentrated using an Amicon Ultra device (4 ml, 10 kDa, Merck Millipore, Darmstadt, Germany) according to the manufacture’s protocol. From a 1.2-l culture, ca. 0.5 ml of the SdnJ solution (13–17 μM) was obtained.
GDP-6-deoxy-d-altrose (final 0.5 mM) and sordaricin (final 0.2 mM, DMSO solution) were mixed with SdnJ (final 14.3 μM, 100 μL), and incubated at 28 °C for 48 h. The reaction was quenched by adding 1 μl of 1 M HCl, and extracted with 200 μl of EtOAc. The organic layer was concentrated using a centrifugal evaporator and the residue was dissolved in 50 μl of CH3OH. Samples were then analyzed using a liquid chromatography–electrospray ionization–tandem mass spectrometry instrument (LCMS 2010EV; Shimadzu) equipped with a LC-20AD × 2 (pump), CTO-20A (column oven), SIL-20A (auto sampler), SPD-20A (UV-Vis detector) and a TSK-Gel ODS100Z (2.0 × 150 mm column, TOSOH, Tokyo, Japan) with a linear gradient of solvents A (water) and B (acetonitrile) as follows: 0–15 min, 40–100% B linear gradient; 15–25 min, 100% B. The flow rate was 0.1 ml min−1. To find the optimum temperature, the reaction was conducted at 16–37 °C. To find the optimum pH, the reaction was conducted in Tris buffer at pHs 7–9. In addition to GDP-6-deoxy-d-altrose, ADP-6-deoxy-d-altrose, TDP-6-deoxy-d-altrose, GDP-d-mannose and GDP-d-glucose were tested as glycosyl donors (0.5 mM each).
References
Hauser, D. & Sigg, H. P. Isolation and decomposition of sordarin. Helv. Chim. Acta 54, 1178–1190 (1971).
Dominguez, J. M. et al. Sordarins: a new class of antifungals with selective inhibition of the protein synthesis elongation cycle in yeasts. Antimicrob. Agents Chemother. 42, 2274–2278 (1998).
Soe, R. et al. Sordarin derivatives induce a novel conformation of the yeast ribosome translocation factor eEF2. J. Biol. Chem. 282, 657–666 (2007).
Hanadate, T. et al. FR290581, a novel sordarin derivative: synthesis and antifungal activity. Bioorg. Med. Chem. Lett. 19, 1465–1468 (2009).
Kamai, Y., Kakuta, M., Shibayama, T., Fukuoka, T. & Kuwahara, S. Antifungal activities of R-135853, a sordarin derivative, in experimental candidiasis in mice. Antimicrob. Agents Chemother. 49, 52–56 (2005).
Bueno, J. M., Chicharro, J., Fiandor, J. M., Gomez de las Heras, F. & Huss, S. Antifungal Sordarins. Part 4: synthesis and structure—activity relationships of 3',4'-fused alkyl-tetrahydrofuran derivatives. Bioorg. Med. Chem. Lett. 12, 1697–1700 (2002).
Castro, J. et al. Antifungal sordarins. part 3: synthesis and structure-activity relationships of 2',3'-fused oxirane derivatives. Bioorg. Med. Chem. Lett. 12, 1371–1374 (2002).
Arribas, E. M. et al. Antifungal sordarins. Synthesis and structure-activity relationships of 3'-O-substituted derivatives. Bioorg. Med. Chem. Lett. 12, 117–120 (2002).
Bueno, J. M., Cuevas, J. C., Fiandor, J. M., Garcia-Ochoa, S. & Gomez de las Heras, F. Antifungal sordarins. Synthesis and structure-activity relationships of 3',4'-fused dioxolane and dioxane derivatives. Bioorg. Med. Chem. Lett. 12, 121–124 (2002).
Martinez, A., Regadera, J., Jimenez, E., Santos, I. & Gargallo-Viola, D. Antifungal efficacy of GM237354, a sordarin derivative, in experimental oral candidiasis in immunosuppressed rats. Antimicrob. Agents Chemother. 45, 1008–1013 (2001).
Martinez, A. et al. Antifungal activities of two new azasordarins, GW471552 and GW471558, in experimental models of oral and vulvovaginal candidiasis in immunosuppressed rats. Antimicrob. Agents Chemother. 45, 3304–3309 (2001).
Herreros, E., Almela, M. J., Lozano, S., Gomez de las Heras, F. & Gargallo-Viola, D. Antifungal activities and cytotoxicity studies of six new azasordarins. Antimicrob. Agents Chemother. 45, 3132–3139 (2001).
Quesnelle, C. A. et al. Sordaricin antifungal agents. Bioorg. Med. Chem. Lett. 13, 519–524 (2003).
Serrano-Wu, M. H. et al. Oxime derivatives of sordaricin as potent antifungal agents. Bioorg. Med. Chem. Lett. 12, 943–946 (2002).
Regueiro-Ren, A. et al. Core-modified sordaricin derivatives: synthesis and antifungal activity. Bioorg. Med. Chem. Lett. 12, 3403–3405 (2002).
Kaneko, S. et al. Synthesis of Sordaricin analogues as potent antifungal agents against Candida albicans . Bioorg. Med. Chem. Lett. 12, 803–806 (2002).
Arai, M., Harasaki, T., Fukuoka, T., Kaneko, S. & Konosu, T. Synthesis and evaluation of novel pyrrolidinyl sordaricin derivatives as antifungal agents. Bioorg. Med. Chem. Lett. 12, 2733–2736 (2002).
Cuevas, J. C., Lavandera, J. L. & Martos, J. L. Design and synthesis of simplified sordaricin derivatives as inhibitors of fungal protein synthesis. Bioorg. Med. Chem. Lett. 9, 103–108 (1999).
Tse, B. et al. Alkyl side-chain derivatives of sordaricin as potent antifungal agents against yeast. Bioorg. Med. Chem. Lett. 8, 2269–2272 (1998).
Chakraborty, B., Mukherjee, R. & Sengupta, J. Structural insights into the mechanism of translational inhibition by the fungicide sordarin. J. Comput. Aided Mol. Des. 27, 173–184 (2013).
Schule, A., Liang, H., Vors, J. P. & Ciufolini, M. A. Synthetic studies toward sordarin: building blocks for the terpenoid core and for analogues thereof. J. Org. Chem. 74, 1587–1597 (2009).
Liang, H. Sordarin, an antifungal agent with a unique mode of action. Beilstein J. Org. Chem. 4, 31 (2008).
Liang, H., Schule, A., Vors, J. P. & Ciufolini, M. A. An avenue to the sordarin core adaptable to analog synthesis. Org. Lett. 9, 4119–4122 (2007).
Chiba, S., Kitamura, M. & Narasaka, K. Synthesis of (-)-sordarin. J. Am. Chem. Soc. 128, 6931–6937 (2006).
Borschberg, H.-J. Zürich, E. T. H. Zur Biogenese des Sordarins (1975) PhD Dissertation.
Toyomasu, T. et al. Fusicoccins are biosynthesized by an unusual chimera diterpene synthase in fungi. Proc. Natl Acad. Sci. USA 104, 3084–3088 (2007).
Kim, S. Y. et al. Cloning and heterologous expression of the cyclooctatin biosynthetic gene cluster afford a diterpene cyclase and two p450 hydroxylases. Chem. Biol. 16, 736–743 (2009).
Meguro, A. et al. An unusual terpene cyclization mechanism involving a carbon-carbon bond rearrangement. Angew. Chem. Int. Ed. 54, 4353–4356 (2015).
Janke, R., Gorner, C., Hirte, M., Bruck, T. & Loll, B. The first structure of a bacterial diterpene cyclase: CotB2. Acta Crystallogr. D Biol. Crystallogr. 70, 1528–1537 (2014).
Jeny, L. & Borschberg, H.-J. Synthesis of the dolabellane diterpene hydrocarbon (±)-δ-araneosene. Helv. Chim. Acta 78, 715–731 (1995).
Mander, L. N. & Thomson, R. J. Total synthesis of sordaricin. J. Org. Chem. 70, 1654–1670 (2005).
Mander, L. N. & Thomson, R. J. Total synthesis of sordaricin. Org. Lett. 5, 1321–1324 (2003).
Kato, N., Kusakabe, S., Wu, X., Kamitamari, M. & Takeshita, H. Total synthesis of optically active sordaricin methyl ester and its Δ2-derivative. Chem. Commun. 1002–1004 (1993).
Mander, L. N. & Robinson, R. P. Studies on the synthesis of the diterpenoid mold metabolite sordaricin. Exploration of a prospective biogenetic intramolecular [4+2] cycloaddition. J. Org. Chem. 56, 3595–3601 (1991).
Daferner, M., Mensch, S., Anke, T. & Sterner, O. Hypoxysordarin, a new sordarin derivative from Hypoxylon croceum . Z. Naturforsch. C 54, 474–480 (1999).
Kato, N., Tanaka, S. & Takeshita, H. Total synthesis of cycloaraneosene, a fundamental hydrocarbon of epi-fusicoccane diterpenoids, and the structure revision of its congener, hydroxycycloaraneosene. Chem. Lett. 15, 1989–1992 (1986).
Kato, N., Wu, X., Tanaka, S. & Takeshita, H. The total synthesis of 8β-hydroxycycloaraneosene by means of an eight-membered ring-formation via a Lewis acid-catalyzed ene reaction. Confirmation of its natural occurrence. Bull. Chem. Soc. Japan 63, 1729–1734 (1990).
Galagan, J. E. et al. The genome sequence of the filamentous fungus Neurospora crassa . Nature 422, 859–868 (2003).
Nowrousian, M. et al. De novo assembly of a 40Mb eukaryotic genome from short sequence reads: Sordaria macrospora, a model organism for fungal morphogenesis. PLoS Genet 6, e1000891 (2010).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucl. Acids Res 33, W465–W467 (2005).
Toyomasu, T. et al. Identification of diterpene biosynthetic gene clusters and functional analysis of labdane-related diterpene cyclases in Phomopsis amygdali. Biosci. Biotechnol. Biochem. 72, 1038–1047 (2008).
Christianson, D. W. Structural biology and chemistry of the terpenoid cyclases. Chem. Rev. 106, 3412–3442 (2006).
Kim, H. J. et al. Biosynthesis of spinosyn in Saccharopolyspora spinosa: synthesis of permethylated rhamnose and characterization of the functions of SpnH, SpnI, and SpnK. J. Am. Chem. Soc. 132, 2901–2903 (2010).
Kreuzman, A. J., Turner, J. R. & Yeh, W. K. Two distinctive O-methyltransferases catalyzing penultimate and terminal reactions of macrolide antibiotic (tylosin) biosynthesis. Substrate specificity, enzyme inhibition, and kinetic mechanism. J. Biol. Chem. 263, 15626–15633 (1988).
Li, S., Anzai, Y., Kinoshita, K., Kato, F. & Sherman, D. H. Functional analysis of MycE and MycF, two O-methyltransferases involved in the biosynthesis of mycinamicin macrolide antibiotics. Chembiochem 10, 1297–1301 (2009).
Patallo, E. P. et al. Deoxysugar methylation during biosynthesis of the antitumor polyketide elloramycin by Streptomyces olivaceus. Characterization of three methyltransferase genes. J. Biol. Chem. 276, 18765–18774 (2001).
Paulus, T. J. et al. Mutation and cloning of eryG, the structural gene for erythromycin O-methyltransferase from Saccharopolyspora erythraea, and expression of eryG in Escherichia coli . J. Bacteriol. 172, 2541–2546 (1990).
Rodriguez, L. et al. Functional analysis of OleY L-oleandrosyl 3-O-methyltransferase of the oleandomycin biosynthetic pathway in Streptomyces antibioticus . J. Bacteriol. 183, 5358–5363 (2001).
Kudo, F., Yonezawa, T., Komatsubara, A., Mizoue, K. & Eguchi, T. Cloning of the biosynthetic gene cluster for naphthoxanthene antibiotic FD-594 from Streptomyces sp. TA-0256. J. Antibiot. 64, 123–132 (2011).
Mulichak, A. M., Bonin, C. P., Reiter, W. D. & Garavito, R. M. Structure of the MUR1 GDP-mannose 4,6-dehydratase from Arabidopsis thaliana: implications for ligand binding and specificity. Biochemistry. 41, 15578–15589 (2002).
Vogan, E. M. et al. Crystal structure at 1.8Å resolution of CDP-D-glucose 4,6-dehydratase from Yersinia pseudotuberculosis . Biochemistry 43, 3057–3067 (2004).
Webb, N. A., Mulichak, A. M., Lam, J. S., Rocchetta, H. L. & Garavito, R. M. Crystal structure of a tetrameric GDP-D-mannose 4,6-dehydratase from a bacterial GDP-D-rhamnose biosynthetic pathway. Protein Sci. 13, 529–539 (2004).
Medema, M. H. & Fischbach, M. A. Computational approaches to natural product discovery. Nat. Chem. Biol. 11, 639–648 (2015).
Wiemann, P. & Keller, N. P. Strategies for mining fungal natural products. J. Ind. Microbiol. Biotechnol. 41, 301–313 (2014).
Bachmann, B. O., Van Lanen, S. G. & Baltz, R. H. Microbial genome mining for accelerated natural products discovery: is a renaissance in the making? J. Ind. Microbiol. Biotechnol. 41, 175–184 (2014).
Blanco-Ulate, B., Rolshausen, P. E. & Cantu, D. Draft genome sequence of the grapevine dieback fungus Eutypa lata UCR-EL1. Genome Announc. 1, e00228–13 (2013).
Baroncelli, R. et al. Draft whole-genome sequence of the biocontrol agent Trichoderma harzianum T6776. Genome Announc. 3, e00647–15 (2015).
Vicente, F. et al. Distribution of the antifungal agents sordarins across filamentous fungi. Mycol. Res. 113, 754–770 (2009).
Jiang, J. et al. Genome mining in Streptomyces avermitilis: a biochemical Baeyer-Villiger reaction and discovery of a new branch of the pentalenolactone family tree. Biochemistry 48, 6431–6440 (2009).
Seo, M. J., Zhu, D., Endo, S., Ikeda, H. & Cane, D. E. Genome mining in Streptomyces. Elucidation of the role of Baeyer-Villiger monooxygenases and non-heme iron-dependent dehydrogenase/oxygenases in the final steps of the biosynthesis of pentalenolactone and neopentalenolactone. Biochemistry 50, 1739–1754 (2011).
Franceschini, S. et al. Exploring the structural basis of substrate preferences in Baeyer-Villiger monooxygenases: insight from steroid monooxygenase. J. Biol. Chem. 287, 22626–22634 (2012).
Yachnin, B. J., Sprules, T., McEvoy, M. B., Lau, P. C. & Berghuis, A. M. The substrate-bound crystal structure of a Baeyer-Villiger monooxygenase exhibits a Criegee-like conformation. J. Am. Chem. Soc. 134, 7788–7795 (2012).
Kasahara, K. et al. Solanapyrone synthase, a possible Diels-Alderase and iterative type I polyketide synthase encoded in a biosynthetic gene cluster from Alternaria solani . Chembiochem 11, 1245–1252 (2010).
Hashimoto, T. et al. Biosynthesis of versipelostatin: identification of an enzyme-catalyzed [4+2]-cycloaddition required for macrocyclization of spirotetronate-containing polyketides. J. Am. Chem. Soc. 137, 572–575 (2015).
Kim, H. J., Ruszczycky, M. W. & Liu, H. W. Current developments and challenges in the search for a naturally selected Diels-Alderase. Curr. Opin. Chem. Biol. 16, 124–131 (2012).
Becker, D. J. & Lowe, J. B. Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R–53R (2003).
Sanz, S. et al. Biosynthesis of GDP-fucose and other sugar nucleotides in the blood stages of Plasmodium falciparum . J. Biol. Chem. 288, 16506–16517 (2013).
Lau, S. T. & Tanner, M. E. Mechanism and active site residues of GDP-fucose synthase. J. Am. Chem. Soc. 130, 17593–17602 (2008).
Zhou, H. et al. The crystal structure of human GDP-L-fucose synthase. Acta Biochim. Biophys. Sin 45, 720–725 (2013).
Hall, R. M. et al. The production of novel sordarin analogues by biotransformation. J. Antibiot. 54, 948–957 (2001).
Tsunematsu, Y. et al. Elucidation of pseurotin biosynthetic pathway points to trans-acting C-methyltransferase: generation of chemical diversity. Angew. Chem. Int. Ed. 53, 8475–8479 (2014).
Hansen, F. T. et al. An update to polyketide synthase and non-ribosomal synthetase genes and nomenclature in Fusarium. Fungal Genet. Biol. 75, 20–29 (2015).
Campbell, C. D. & Vederas, J. C. Biosynthesis of lovastatin and related metabolites formed by fungal iterative PKS enzymes. Biopolymers 93, 755–763 (2010).
Brown, D. W., Butchko, R. A., Baker, S. E. & Proctor, R. H. Phylogenomic and functional domain analysis of polyketide synthases in Fusarium. Fungal Biol 116, 318–331 (2012).
Awakawa, T. et al. Physically discrete beta-lactamase-type thioesterase catalyzes product release in atrochrysone synthesis by iterative type I polyketide synthase. Chem. Biol. 16, 613–623 (2009).
Brock, N. L., Huss, K., Tudzynski, B. & Dickschat, J. S. Genetic dissection of sesquiterpene biosynthesis by Fusarium fujikuroi . Chembiochem 14, 311–315 (2013).
Gloster, T. M. Advances in understanding glycosyltransferases from a structural perspective. Curr. Opin. Struct. Biol. 28, 131–141 (2014).
Davisson, V. J. et al. Phosphorylation of isoprenoid alcohols. J. Org. Chem. 51, 4768–4779 (1986).
Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research in Innovative Area 22108003 from the Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Dedicated to Professor David E. Cane for his contribution to biosynthesis studies of antibiotics.
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Kudo, F., Matsuura, Y., Hayashi, T. et al. Genome mining of the sordarin biosynthetic gene cluster from Sordaria araneosa Cain ATCC 36386: characterization of cycloaraneosene synthase and GDP-6-deoxyaltrose transferase. J Antibiot 69, 541–548 (2016). https://doi.org/10.1038/ja.2016.40
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2016.40
This article is cited by
-
Efficient exploration of terpenoid biosynthetic gene clusters in filamentous fungi
Nature Catalysis (2022)
-
Discovery and characterization of a terpene biosynthetic pathway featuring a norbornene-forming Diels-Alderase
Nature Communications (2022)
-
Diversity of biologically active secondary metabolites in the ascomycete order Sordariales
Mycological Progress (2022)
-
Synthaser: a CD-Search enabled Python toolkit for analysing domain architecture of fungal secondary metabolite megasynth(et)ases
Fungal Biology and Biotechnology (2021)
