
Abstract
The piericidin family of microbial metabolites features a 4-pyridinol core linked with a methylated polyketide side chain. Piericidins are exclusively produced by actinomycetes, especially members of the genus Streptomyces. The close structural similarity with coenzyme Q renders the piericidins important NADH-ubiquinone oxidoreductase (complex I) inhibitors in the mitochondrial electron transport chain. Because of the significant activities of the piericidins, which include insecticidal, antimicrobial and antitumor effects, total syntheses of the piericidins were developed using various synthetic strategies. The biosynthetic origin of this class has also been the subject of investigation. This review covers the isolation and structure determination of the natural piericidins, their chemical modification, the total syntheses of natural and unnatural analogs, their biosynthesis, and reported biological activities together with structure–activity relationships. Given the fundamental biology of this class of metabolites, the piericidin family will likely continue to attract attention as biological probes of important biosynthetic processes.
Similar content being viewed by others


Introduction
The piericidins are a family of structurally related compounds isolated from actinomycetes, especially from members of the genus Streptomyces. The striking structural resemblance between piericidins (such as piericidin A, 1) and coenzyme Q (2), shown in Figure 1, suggested that piericidins act as coenzyme Q antagonists. This has been confirmed by the finding that piericidins are specific and potent NADH-ubiquinone oxidoreductase (complex I) inhibitors in the mitochondrial electron transport chain.1 Yoshida and Takahashi2 reviewed the piericidins in 1978, before the full complexity of this class was known.

Structure of piericidin A (1) and the coenzyme Q complex (2).
With the development of new chemical diversity and enhanced biological studies of the piericidins during the past four decades, this unique class is showing potential value in cell biology studies, although the toxicities to mammals are still inescapable. In this review, we examine the up-to-date literature on both natural and synthetic piericidins, and include more recent studies defining their total syntheses and biosynthesis. Information on bioactivities and structure–activity relationships (SAR) is also discussed.
The Origins of the Piericidins
Piericidin A (also named piericidin A1 in some references) was reported by Takahashi and co-workers as a new insecticidal metabolite produced by cultures of the soil-derived actinomycete Streptomyces mobaraensis. The original reports included details of its isolation, physiological activity, functional groups and a partial structure.3, 4, 5 The structure of piericidin A was originally proposed in 1965 as 1a (Figure 1) on the basis of extensive degradation studies and analysis of the 1H NMR data.6 Piericidin A resembles coenzyme Q in its overall structure containing a pyridine ring with two adjacent methoxy groups, rather than the identically substituted p-benzoquinone functionality.7 The structure of the pyridine ring was subsequently confirmed by synthesis,8 and the configurations of the asymmetric centers, C-9 and C-10, were proposed to be S in 1967.9, 10
Ultimately, by a combination of 13C-NMR spectral analyses and 13C feeding experiments, the structure of piericidin A (1) was revised by reassigning the C-4–C-5 olefin to C-5–C-6. Further, in 1977, the same group confirmed their assignment by selective reduction and the application of mass spectral analysis.11, 12 Then, in 1983, the configurations of C-9 and C-10 were revised to 9R, 10R on the basis of strong optical rotation data.13 The full stereochemistry of the side chain was also confirmed by enantioselective synthesis several years later yielding solid proof of the structure of piericidin A (1).14, 15
Natural piericidin aglycones
Over time, numerous new piericidin analogs were discovered. Piericidin B (3, also named piericidin B1 in some references) was also originally observed in the cultivation of Streptomyces mobaraensis in 1963.3 Piericidin B was assigned as the C-10 methoxy analog of A, apparently produced during fermentation and work-up.9
Sometime later, Takahashi reported that 14 new piericidins could be extracted from the cultured mycelium of Streptomyces pactum.16 These piericidins were classified into four groups, piericidins A (1), A2–A4 (4–6), B (3), B2–B4 (7–9), C1–C4 (10–13) and D1–D4 (14–17), according to their chromatographic behavior, and their structures were assigned on the basis of mass, 1H and 13C NMR spectral analyses (Figure 2).16 The UV spectra of these piericidins are classified into two types: one, due to piericidins having an odd suffix, shows λmax at 239 nm, and the other, due to even suffixed piericidins, shows λmax at 225 nm.17

Structures of piericidin aglycones 1 and 3–17.
Two N-oxide derivatives of piericidin, piericidin B1 N-oxide (18) and piericidin B5 N-oxide (19), together with piericidin B5 (20), were isolated from cultures of Streptomyces strain MJ288-OF3. The structures of the new N-oxides were determined by NMR and high-resolution MS analyses and confirmed by zinc powder reduction to yield piericidin B (3) and piericidin B5 (20).18, 19
Other piericidin aglycones that include Mer-A2026 A (21) and B (22), which lack the methoxy group at C-3′ (numbered as C-5′ in the original reference), were isolated from cultures of Streptomyces pactum Me2108. Two new piericidins, IT-143-A (23) and B (24) were isolated from the culture mycelia of Streptomyces sp. IT-143.20 A new member of the piericidin family, JBIR-02 (25), was isolated from the mycelium of Streptomyces sp. ML55, together with two known piericidin derivatives, piericidin A (1) and IT-143-B (24).21 NMR analysis, including NOE experiments, showed that 25 was a 3:1 tautomeric mixture of pyridone (25a) and hydroxypyridine (25b). The absolute configurations of 22 (9R,10R) and 25 (9R,10R) were subsequently established by total synthesis.22
Piericidin C5 (26), together with piericidins C1 (10), C3 (12), D1 (14) and A3 (5), were isolated from the fermentation broth of a Streptomyces sp., whereas piericidin C6 (27), as well as piericidins C2 (11) and C4 (13) were isolated from the culture of a Nocardioides species. This is the first report of piericidins produced by a microorganism other than members of the genus Streptomyces.23 Piericidins C7 (28) and C8 (29) were found to be produced by a Streptomyces sp., associated with a marine ascidian.24, 25 7-Demethylpiericidin A1 (30) was isolated from a soil-derived Streptomyces sp. SN-198.26 A deep sea-derived Streptomyces sp. SCSIO 03032, yielded piericidin E1 (31) a novel analog featuring a C-2/C-3 epoxide ring (Figure 3).27

Structures of piericidin aglycones 18–31.
Natural piericidin glycosides
The first piericidin glycosides, glucopiericidins A (32) and B (33) were reported in 1987. These two glycosides, defined as piericidin A, 10-O-β-d-glucoside (glucopiericidin A, 32) and piericidin A, 4′-O-β-d-glucoside (glucopiericidin B, 33), were isolated from the culture broth of the soil-derived Streptomyces pactum S48727, together with the aglycone piericidin A (1).28 In the cultivation, glucopiericidin A (32) was produced after the appearance of piericidin A (1), indicating that glucopiericidin A (32) was produced by glucosylation of piericidin A (1). Glucopiericidin A (32) was a main product after 96-h incubation, whereas piericidin A (1) and glucopiericidin B (33) were minor.28
Another two new piericidin glycosides, glucopiericidinols A1 (34) and A2 (35), possessing an additional hydroxy group at C-3, were isolated from the cultured broth of Streptomyces sp. OM-5689.29 In the same strain, another glycoside called 13-hydroxyglucopiericidin A (36) was reported in the next year.30 Two new piericidin rhamnosides, 4′-rhamnopiericidin A1 (37) and 7-demethyl-4′-rhamnopiericidin A1 (38), were isolated from the soil Streptomyces sp. SN-198 and reported in 1990 and 1996, respectively.26, 31 In the mycelial extract of Streptomyces sp. DO-100, isolated from a soil sample, a piericidin glycoside with an unusual methylhexose moiety was discovered, and its structure was determined to be 4′-deoxytalopiericidin A1 (39) by one-dimensional NMR and NOE experiments.32 A new piericidin glycoside derivative named glucopiericidin C (40) was isolated, together with glucopiericidin A and piericidin A, from a marine sediment-derived Streptomyces isolate, B8112. The structure of glucopiericidin C (40) was assigned as the 10-O-β-d-glucoside of Mer-A2026A. Glucoside 40 was similar to glucopiericidin A, except for the loss of the methoxy group at C-3′ (Figure 4).33

Structures of piericidin glycosides 32–40.
Chemical modification and total synthesis
Synthetic piericidin analogues
The rare structure of piericidin A consists of a 2,3-dimethoxy-5-methyl-4-pyridinol ring bonded at C-6 with a poly-unsaturated and -methylated side chain. Schmidtchen and Rapoport34 prepared the pyridinol ring and attached conventional polyisoprenoid side chains for structure–activity studies. The synthetic piericidin analogues (41a–e) were tested for their activities of complex I inhibition. The farnesyl analogue 41c illustrated potent inhibitory effects, with similar activity to piericidin A (1). Surprisingly, the solanesyl analogue 41e was two orders of magnitude less active.34
Takahashi et al.35 also synthesized various piericidin analogues to clarify the respiratory inhibition relationships (SAR) of piericidins and their analogs.36 Finally, 12 analogues (42a–d, 43a–d and 44a–d), comprising three series depending on the ring structures, were synthesized by a Wittig reaction, as well as their saturated side-chain analogues (45a–d, 46a–d and 47a–d).35
The diacetate 48 and methyl ether 49 derivatives of Mer-A2026A (21, 3′-demethoxyl piericidin A1) were prepared as part of the structure determination of 21.37 Two piericidin analogues, 50 and 51, having a simple benzene ring were synthesized by a palladium-catalyzed cross-coupling reaction and the reaction of sulfone and aldehyde using the Julia coupling procedure.38 Analog 50 with a 2E olefin configuration has the same side chain as natural piericidin (piericidin B, 3) and 51 is the 2Z-isomer of 50 at the side chain (Figure 5).

Structures of synthetic piericidin analogues 41–51.
Following the successful completion of the total synthesis of piericidin A (1) and B1 (3), Boger and co-workers also prepared a series of key analogues.39 Ent-Piericidin A1 (52) and the C-10 hydroxy diastereomer (53) were prepared from the pivalate ester used as an intermediate in the total synthesis of 3 and its ketone analog 41. The C-4′-acetate, C-10 ketone 54 was prepared from the alcohol 55, which was derived from piericidin A (1). Mild acetate hydrolysis of 54 provided the C-10 ketone 56. The C-4′ methyl ethers 57 and 58 were prepared by selective methylation of 1 and 41c, another analog they also obtained. More significantly, C-4′-deshydroxypiericidin A1 (59) and the C-4′-deshydroxy analogue (60) were prepared by total synthesis for biological examination.
Boger and co-workers were also successful in accessing the C-5′-desmethyl and C-4′-deshydroxy–C-5′-desmethyl analogues bearing the natural piericidin side chain as well as the simplified farnesyl side chain, 5′-desmethylpiericidin A1 (61), 4′-deshydroxy-5′-desmethylpiericidin A1 (62) and the corresponding farnesyl side-chain bearing analogues (63, 64; Figure 6).

Structures of synthetic piericidin analogues 52–64.
Total synthesis of piericidins
Although the preparation of the fully elaborated pyridine ring system substituted with simplified side chains was reported in 1977,34 as well as an asymmetric synthesis of the C-6–C-13 segment of the side chain in 1991,15 there were no total syntheses of piericidins until 2005. Boger and co-workers reported the first total synthesis of piericidin A (1) and B1 (3) in 2005 in an early paper,40 and then provided full details in the next year.39 Shortly following Boger’s preliminary disclosure, Keaton and Phillips41 reported a complementary total synthesis of 7-demethylpiericidin A1 (30), a natural product closely related to piericidin A (1). In contrast to Boger’s method, Akita et al.42, 43 reported a new total synthesis, in 2009, of 1 and 3 based on a convergent route involving a Julia coupling strategy. Lipshutz and Amorelli44 also described a unique strategy for the key disconnection, highlighting a modified Negishi carboalumination/Ni-catalyzed cross-coupling on a polyenyne precursor. Subsequently, Gademann and co-workers reported the total syntheses of the piericidin-related natural products Mer-A2026B (22) and JBIR-02 (25).22
Central to Boger’s approach is an inverse electron demand Diels–Alder reaction between an N-sulfonyl-1-aza-1,3-butadiene and tetramethoxyethene followed by Lewis acid promoted aromatization to assemble the functionalized pyridine core. Additional key elements in the convergent approach include the use of an asymmetric anti-aldol reaction to install the relative and absolute stereochemistry at C-9 and C-10, a modified Julia Olefination for formation of the C-5–C-6 trans-double bond with convergent assemblage of the side chain, and a penultimate heterobenzylic Stille cross-coupling reaction of the pyridyl core with the fully elaborated side chain (Figure 7).39, 40

Boger’s synthetic strategy for piericidins A (1) and B1 (3).
Shortly after Boger’s report on the synthesis of piericidin A (1) and B1 (3), Phillips and Keaton41 reported a convergent total synthesis of 7-demethylpiericidin A1 (30) that is patterned on the strategy outlined in Figure 8. The total synthesis proceeded in nine steps from Tiglic aldehyde, and features an application of Titanium (II)-mediated cyclization of a (silyloxy)enyne as the key reaction for the assembly of the polyene domain.41

Phillips’ synthetic strategy for 7-demethylpiericidin A1 (30).
Akita et al. reported the synthesis of a (±)-piericidin B (3) analog possessing a benzene ring instead of pyridine ring in 1997.45 Further, in 2009, they reported a new total synthesis of piericidin A (1) and B1 (3) based on a convergent synthesis via Julia coupling different than Boger’s method, which is based on carbon–carbon bond formation by Stille cross-coupling and the construction of the two stereogenic centers in the side chain by the use of an asymmetric anti-aldol reaction.39, 40 Akita’s synthetic plan for 1 and 3 was based on double bond formation between the left and right halves as shown in Figure 9. The construction of the two stereogenic centers in the right half could be achieved by lipase-catalyzed enantioselective hydrolysis of α-methyl-β-acetoxy ester. The non-conjugated aldehyde left half corresponding to the C-1–C-5 unit of the piericidin side chain was prepared starting from the known allyl alcohol congener.42, 43

Akita’s synthetic strategy for piericidins A (1) and B1 (3).
Lipschitz’s and Amorelli’s approach to a practical synthesis of piericidin A (1) highlights a modified Negishi carboalumination followed by a Ni-catalyzed cross-coupling strategy. This strategy allows for couplings of benzylic chlorides and in situ generated vinylalanes, arrived at via stereoselective carboalumination of terminal alkynes. Retrosynthetically, the key disconnection features a penultimate one-pot Ni-catalyzed coupling of vinylalane (right half), generated in situ via a modified carboalumination, to the chloromethylated pyridine (left half, Figure 10). The skipped enyne is anticipated by a propargyl selective (over allenyl) coupling of a corresponding vinyl iodide and TMS-propyne. A vinylogous Mukaiyama aldol reaction generated the eight-carbon vicinal methyl/hydroxyl side chain framework.44 This route, to the synthesis of piericidin A (1) in a total of 18 steps from commercial material, involves a longest linear sequence of 11 steps from the N,O-silyl ketene acetal (Figure 10), which overall compares very favorably with the previous synthesis of piericidin A (1).39, 40, 41

Lipschutz’s synthetic strategy for piericidin A (1).
The total syntheses of the piericidin related natural products Mer-A2026B (22) and JBIR-02 (25) were reported in 2013 by Gademann’s group.22 The total synthesis started with an Ir-catalyzed one-pot C–H activation reaction in combination with an oxidation procedure for the preparation of the protected bromopyridinol in five steps. The preparation of the side chain was also started from the N,O-silyl ketene acetal by a highly diastereoselective vinylogous Mukaiyama aldol reaction. The final connection of both moieties, the 4-hydroxypyridine core and the fully elaborated polyene chain, was realized after extensive experimentation with the Negishi cross-coupling reaction without isomerization of the labile side chain. Furthermore, this strategy opens up the stage for the synthesis of a number of piericidins by simply changing the coupling partner for the Horner–Wadsworth–Emmons reaction or the organometallic species for the cross-coupling reaction. In brief, key features of the synthetic approach involved a C–H activation/oxidation procedure for the preparation of the highly functionalized pyridine moiety, a vinylogous Mukaiyama aldol reaction, and a crucial Negishi cross-coupling of an advanced pyridine derivative with an allylic side-chain precursor (Figure 11).22

Key features of Gademann’s total syntheses of Mer-A2026B (22) and JBIR-02 (25).
Biosynthsis of piericidin A
In 1969, Takahashi and co-workers46 had proven by degradation studies that the long-branched C23 chain of the piericidins (65 in Figure 12) was formed from five propionate and four acetate units, and that a nitrogen atom was introduced at the terminal part of the chain, followed by cyclization to form the pyridine ring. The two methoxyl groups on the pyridine rings of the piericidins were derived from S-methylmethionine.46 This conclusion had been confirmed by Tanabe and Seto47 in the next year by NMR measurements of satellite bands due to sp3-carbons, which were readily assigned by a selective 13C–1H decoupling technique. Although the total synthesis of piericidin A has been achieved successfully, studies on the biosynthesis of piericidins were limited before 2012.

Long-branched C23 chain (65) in the biosynthesis of piericidins.
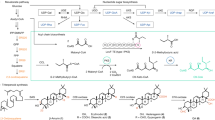
In 2012, the You and Tang groups provided an overview of the biosynthetic pathway for piericidin A (1; Figure 13).48 Sequential analysis of the Streptomyces piomogeues genome revealed six modular polyketide synthases, an amidotransferase, two methyltransferases and a monooxygenase responsible for the production of 1. Gene functional analysis and deletion studies revealed with confidence that 1 originates from a modular polyketide synthase pathway.48 Unexpectedly, the nitrogen atom in piericidin A was found to be a result of an ATP-dependent amidotransferase rather than the more common hybrid polyketide synthase–non ribosomal peptide synthetase pathway.48

Proposed biosynthetic pathway of piericidin A (1).48
The piericidin A (1) gene cluster was also identified from the deep-sea-derived Streptomyces sp. SCSIO 03032 by Zhang and colleagues in 2014.27 In vivo and in vitro experiments verified PieE as a 4′-hydroxylase and PieB2 as a 4′-O-methyltransferase (sic., the C number 4′ is according to the original reference), allowing the elucidation of the post-polyketide synthase modification steps involved in piericidin A biosynthesis (Figure 14).27

Genetic organization of the pie gene cluster in Streptomyces sp. SCSIO 03032 and the proposed piericidin A (1) biosynthetic pathway.27
Piericidin bioactivities
Complex I inhibitory activities and SAR studies
Piericidin A (1) is well known as an effective inhibitor of the electron transport chain, also called the respiratory chain.49, 50 There are four protein complexes, labeled complex I–IV, in the electron transport chain, which are involved in moving electrons from NADH and succinate to molecular oxygen. Piericidin A (1) is an especially potent inhibitor of complex I that bind to its ubiquinone binding site,51, 52, 53 with a Ki ranging between 0.654 and 1 nm.55 Respiratory inhibition by piericidin A (1) can be reversed by the addition of vitamin-K3 (menadione) to the inhibited respiratory chain in mammalian mitochondria, but not in insect mitochondria.56
SAR studies of complex I inhibitors are important not only to elucidate the structural factors required for their inhibitory action but also to determine the structural properties of the ubiquinone reduction site in the enzyme. Natural piericidins, as well as synthetic analogues, were examined in SAR studies by Takahashi and co-workers with mammalian and insect mitochondria, in and before 1980.6, 11, 16, 35, 36 Several review papers and book chapters also cited their results.36, 57, 58, 59 In 2006, Boger also reported detailed SAR studies using synthetic piericidin analogues measuring their complex I inhibitory activities.39
On the basis of SAR studies of a number of synthetic piericidin analogues possessing different side chains, it was concluded that a branched methyl group at C-3 and unsaturation between C-2 and C-3 are important for complex I inhibitory activity.35, 36 It was considered that the side chain might serve primarily to enhance the hydrophobicity of the whole structure, as derivatives possessing a simple long alkyl chain retained fairly potent activity.60 However, the location of the hydrophobic side chain at either the C-5′ or C-6′ position of the pyridine ring does not affect the inhibitory potency.36, 60 The phenolic hydroxy group on the pyridine ring was seen as necessary to maintain the high level of activity.61 Takahashi and Yoshida synthesized a variety of piericidin analogues to elucidate the contributions of the C-2′, C-3′ and C-5′ functional groups on the pyridine ring, but their assay results were somewhat confusing.35, 36 The Boger group examined 15 synthetic piericidin analogues (41a, 41c and 52–64), together with natural piericidin A (1), B1 (3) and rotenone, for inhibition of complex I.39 The results showed that small modifications in the side chain had little impact on this activity; O-methylation of the C-10 hydroxy group (3, half-maximal inhibitory concentration (IC50) 5.1 nm) with piericidin A (1, IC50 3.7 nm), its conversion to a ketone (56, IC50 6.3 nM), and inversion of the C-10-OH configuration (53, IC50 8 nM) resulted in very small changes in potency. The piericidin analog with the simplified farnesyl side chain (41c, IC50 3.8 nm) was similar in potency to 1. However, the overriding importance of the side chain became quite clear in examining 41a (IC50 10 000 nm) in which removal of the C-5–C-16 segment resulted in an extreme loss in activity (Figure 15).

Brief structure–activity relationships of piericidins as complex I inhibitors. A full color version of this figure is available at The Journal of Antibiotics journal online.
The pyridyl C-4′ hydroxyl group also proved to be important for the inhibition of complex I. In acetylated analogs (54, IC50 26 nm; 55, IC50 24 nm) or methylated analogs (57, IC50 130 nm), the transformation led to 7- and 35-fold reductions in potency. Removal altogether as in C4′-deshydroxypiericidin (59, IC50 10 nm) had a surprisingly modest impact. In marked contrast, O-methylation of the C-4′ phenol (58, IC50 750 nm) as well as the removal of the C-4′ phenol (60, IC50 400 nm) in the analogues bearing the simplified farnesyl side chains reduced complex I inhibition >100-fold. These latter studies supported that the C-4′ hydroxy group is central to piericidin′s activity. The C-5′ methyl group of piericidin had a similar remarkable impact on the piericidin activity. Methyl group removal as in 5′-desmethylpiericidin A1 (61, IC50 4.0 nm) had little or no impact on complex I inhibition, whereas its removal in the analogue 63 (IC50 110 nm) bearing the farnesyl side chain reduced complex I inhibition 30-fold relative to either 41c or 1. Just as remarkable, removing both the C-4′ hydroxyl and C-5′ methyl groups as in 62 (IC50 9.0 nm) only reduced the complex I inhibition ca. 2-fold relative to piericidin A itself, whereas these modifications to yield analogue 64, bearing the farnesyl side chain, resulted in a compound that was >500-fold less potent than 1 or 41c. Surprisingly, the potent activity of 59, 61 and especially 62 suggests that the natural product side-chain composition (versus the farnesyl side chain) somehow compensates for losses in binding affinity that result from modifications of the pyridine core (Figure 15).
Boger’s SAR studies showed that the pyridine C-4′ hydroxy group and the C-5′ methyl substituent appear more central to the potency of the piericidins, as their modification or removal leads to reductions in complex I inhibition. Although the side-chain substituents of piericidin A may impact complex I inhibition, this is most significant when key structural features of the pyridine core are removed.
Functions as Complex I inhibitors
Piericidins were first discovered because of their insecticidal activity,3, 62 and mechanistic studies indicated that complex I was the molecular target of piericidins.63 Besides its well-known redox role in the electron transport chain, complex I is also considered to be one of the main production sites of reactive oxygen species (ROS). Complex I inhibitors can be grouped into two classes depend on their effect in the production of ROS. Piericidin A (1), as well as other class A inhibitors such as rotenone, increase ROS production, whereas class B inhibitors (such as stigmatellin, mucidin and capsaicin) appear to directly prevent oxygen reduction presumably acting on the electron escape site.64 In forward electron transfer in complex I, slow superoxide production in the presence of glutamate and malate was enhanced by both rotenone and piericidin A (1).65 Co-exposure of hepatocytes to isoniazid, an anti-tuberculosis drug associated with idiosyncratic liver injury, and nontoxic concentrations of 1 (30 nm) resulted in massive ATP depletion and cell death, indicating that piericidin A (1) triggers isoniazid-induced hepatocellular injury.66
The effect of piericidin A (1) on the sites of inhibition in and around photosystem II was examined, and the results suggested that it binds to cytochrome b6/f complex, out of the photosystem II complex.67 Although mitochondrial complex I inhibition has been linked to sporadic tauopathies, piericidin A (1) was studied to determine whether there is a pathogenic interaction with the P301S mutation. The results showed that 1 significantly increased the number of phospho-tau immunoreactive cells in the cerebral cortex in P301S mice. Furthermore, 1 led to increased levels of pathologically phosphorylated tau only in P301S mice.68 Mitochondrial complex I dysfunction is regarded as underlying dopamine neuron death in Parkinson’s disease models. However, piericidin A (1) as a complex I inhibitor did not induce selective dopamine neuron death.69
Cytotoxic and antimicrobial effects with SAR studies
Piericidin A (1) showed significant cytotoxic activities against several tumor cells, such as mouse leukemia cell line (L1210, IC50 5 nm),39 and was selectively cytotoxic toward human multiple myeloma cells.70 Because of the toxicity of 1 toward cultured bioassay cells, other activities, such as antiviral activity, were not significant or not found.71 In the acute toxicity tests administering piericidin A (1) by i.v. administration in mice, the LD50 value was <1.0 mg kg−1,28, 72 whereas glucopiericidins A (32) and B (33) showed less toxicity with LD50 values between 10 and 30 mg kg−1. Most of the other piericidins, including the glycosides, also showed cytotoxic activities to several cancer cell lines (Table 1).
The piericidin family exhibits an array of antimicrobial activity. The antimicrobial effects of the piericidins against various Gram-positive and Gram-negative bacteria, yeast or fungi are listed in Table 1, although the tested strains differed in their susceptibility to different piericidins. Interestingly, a complex of eight symbiont-produced piericidin derivatives from cocoon extracts of beewolf digger wasps showed that complementary action confers a potent antimicrobial defense.73 The mixture of piericidin antibiotics strongly inhibited all test microbes, and this suggests that beewolf symbionts use a defensive strategy against bacterial and fungal pathogens that corresponds to ‘combination therapy’ or ‘combination prophylaxis’ an approach increasingly used in human medicine.73
Structure–cytotoxic activity relationships of the piericidin aglycones showed that modification or removal of the pyridine C-4′ hydroxy group and the C-5′ methyl substituent could significant reduce cytotoxicity. The side-chain substituents of piericidin A (1) enhanced cytotoxic activity, but their impact seems to be most significant if the pyridine core of 1 is intact.39
The biological activities of the piericidin glycosides are strongly influenced by the type and location of the sugar unit. Glucopiericidin A (32) inhibits the growth of Gram-positive bacteria and the rice blast fungus Piricularia oryzae, whereas glucopiericidin B (33) was more active than 32. Glucopiericidins A-C (32, 33 and 40) showed higher antimicrobial activities than piericidin A (1) or other glycoside analogs.28, 33 It was interesting that the glycosides 3'-rhamnopiericidin A1 (37) and 7-demethyl-3′-rhamnopiericidin A1 (38) showed lower antimicrobial activities against some fungi and Gram-negative bacteria than 7-demethylpiericidin A1 (30) and piericidin A (1). In contrast, 37 and 38 showed stronger cytotoxicity against KB and K562 cells. Among the glycosides, the presence or absence of a methyl group at C-7 did not significantly affect their biological activities.26, 31
Glucopiericidinols A1 (34) and A2 (35) showed almost no activity against various Gram-positive and Gram-negative bacteria, yeast or some fungi, but they did have inhibitory activity against the rice blast fungus Pyricularia oryzae.29 13-Hydroxyglucopiericidin A (36) showed lower activity against P. oryzae, whereas it showed strong cytotoxicity against various tumor cells in vitro. It is noteworthy that 36 showed stronger activity against adriamycin-resistant P388 cells (ADM) than normal P388 cells.30 3′-Deoxytalopiericidin A1 (39) inhibited cancer cells selectively.32 It has been suggested that the differences in these levels of potency and activity may depend on the permeability of the drug to the cell membrane.26
Anti-tumor potency
Phosphatidylinositol (PI) turnover is considered to be correlated with transformation by some types of oncogenes and cellular response to growth factors such as epidermal growth factor (EGF)74 and platelet-derived growth factor (PDGF).75 Piericidin-B1 N-oxide (18) and B5 N-oxide (19) inhibited the EGF-induced PI turnover of A431 cells with an IC50 of 1.2 and 1.1 μg ml−1, respectively, whereas piericidin B (3) and B5 (20) showed weaker inhibitory activity (IC50 5.0 and 10.0 μg ml−1, respectively) than the N-oxides (18 and 19).19, 76 N-oxide 18 showed no inhibitory effects on DNA, RNA or protein synthesis, at the concentration inhibiting PI.77 Glucopiericidin A (32) inhibited PDGF-induced activation of phospholipase-C gamma 1 by reducing the tyrosine kinase activity of the PDGF receptor and it more potently inhibits PI turnover induced by PDGF than by EGF.78
The adenovirus E1A gene product inactivates retinoblastoma tumor suppressor protein (pRB), which has an important role in cell cycle and apoptosis control in mammalian cells, thereby stimulating host cell DNA synthesis.79 Piericidins A, A2, C7, and C8 (1, 4, 28 and 29) showed selective cytotoxicities against rat glia cells transformed with the adenovirus E1A gene (RG-E1A-7) and inhibited the growth of Neuro-2a cells. Mitochondrial dysfunction might be one of the mechanisms causing cell death or growth arrest in certain types of tumors.24
Glucopiericidin A (32) was discovered as a filopodia protrusion inhibitor, but only synergistically with the mitochondrial respiration inhibitor piericidin A (1). It was shown that 32 suppresses glycolysis by functionally targeting the glucose transporter. Simultaneous inhibition of both glycolysis and mitochondrial respiration markedly decreased intracellular ATP levels, indicating that 32 inhibits ATP-dependent filopodia protrusion in carcinoma cells when treated with 1.80
Glucose deprivation causes upregulation of GRP78 and induction of etoposide resistance in human cancer cells, thus downregulating GRP78 expression may be a novel strategy toward anticancer drug development. It was shown that piericidin A (1) suppresses the accumulation of GRP78 protein and was also highly toxic to etoposide-resistant HT-29 cells, with IC50 values for colony formation of 6.4 and 7.7 nm under 2-deoxyglucose supplemented and glucose-deprived conditions, respectively. Interestingly, 1 had no effect under normal growth conditions.81
JBIR-02 (25) was discovered as a regulator of arrestin translocation and was suggested to be a new type of antitumor drug. Compounds 25 and 1 inhibited nuclear export of β-arrestin 2-EYFP (enhanced yellow fluorescent protein) fusion protein in HeLa cells at concentrations of 20 and 40 μm, respectively, whereas IT-143-B (24) had no such effect.21
Animal experiments showed several piericidins could be promising as lead compounds for antitumor drug development. An i.p. injection, at 0.625 mg kg−1, of 3′-deoxytalopiericidin-A1 (39) resulted in the suppression of the growth of colon 26 tumor (T/C=21.8%) implanted s.c. in syngeneic CDF1 mice.32 Piericidin B1 N-oxide (18) reversibly inhibited the growth of A431 cells in situ and suppressed the growth of Ehrlich carcinoma in vivo when administered to mice by i.p. injection.77
Other bioactivities
Studies of piericidin A (1) on the metabolism of isolated adipose cells showed the inhibition of glucose and fructose utilization, and the elimination of the stimulatory effects of proteases on glucose utilization. Piericidin A (1) accelerated glycogenolysis in isolated adipose cells, but exerted no significant effects on the level of lipid content or the oxidation of the cellular components. Lipolysis induced by lipolytic hormones or phosphodiesterase inhibitors, or by both, was also blocked by 1. The effects of 1 on the isolated adipose cells were interpreted as probably being due to the plasma membrane effects and possible effects on the adenylyl (adenyl) cyclase system.82
The effects of 1, glucopiericidins A (32) and B (33) on antibody formation to sheep red blood cells in mouse spleen cell cultures were examined. d-Glucose in the glucopiericidins seemed to be necessary for inhibitory activity, as 1 was less effective than the glucosylated analogs in tests.28 Piericidin A (1) was identified as a selective inhibitor of phagocytosis,83 one of the basic and characteristic properties of macrophages. In addition, 1 was also shown to inhibit the cytokine interleukin-2 production in mouse thymoma EL4 cells, which regulates many essential immune functions.84
Mer-A2026A and B (21 and 22) were found to have potent vasodilating activities.76 The vasodilating effects of 21 and 22 on rat aorta were greater than that of papaverine, a standard vasodilating drug. Mer-A2026A (21; 0.1 mg kg−1), as well as papaverine (1 mg kg−1), showed similar potent depressor effects in spontaneously hypertensive rats, but the duration of the effect caused by 21 was significantly longer than that by papaverine.72
The type III secretion system (T3SS) is a bacterial appendage used by dozens of Gram-negative pathogens to subvert host defenses and cause disease, making it an ideal target for pathogen-specific antimicrobials.85 Compounds 22 and 1 were shown to inhibit Yersinia pseudotuberculosis from triggering T3SS-dependent activation of the host transcription factor NF-κB in HEK293T cells, but were not toxic to mammalian cells at comparable concentrations. Compounds 22 and 1 were described as bacterial T3SS inhibitors less likely to generate resistance.85
Conclusions
The family of piericidin metabolites are some of the most commonly encountered in culture studies of the actinomycetes isolated from soil and marine samples. Forty piericidins, including nine piericidin glycosides, have been isolated to date. The common genus Streptomyces is the main source of the piericidins, with occassional exceptions of piericidin metabolites found produced by Nocardioides species. (Table 1). In those identified Streptomyces strains, S. pactum is a well-known piericidin producer. Although soil-derived actinomycetes have been the predominant source of the piericidins, more recently, marine-derived Streptomyces isolates have shown promise to produce piericidins, especially new piericidin analogs.24, 33
Piericidins are well known as complex I inhibitors. Moreover, some piericidins, such as 3′-deoxytalopiericidin-A1(39)32 and piericidin B1 N-oxide (18), are promising as lead compounds for anti-cancer drug development.76, 77 SAR studies indicate that the sugar component of the piericidin glycosides is important in modulating their physiological activities.33 However, the bioactivities of the piericidin glycosides seem much less well defined and require further refinement, especially regarding their anticancer mechanism of action. Overall, the broad bioactivity of this class of microbial metabolites seems less well understood. Clearly many are potently toxic. Given the potency in a diversity of bioassay systems, the piericidins are likely to captivate interest for some time to come.
References
Hall, C. et al. Piericidin A: a new inhibitor of mitochondrial electron transport. Biochem. Biophys. Res. Commun. 25, 373–377 (1966).
Yoshida, S. & Takahashi, N. Piericidins–naturally occurring inhibitors against mitochondrial respiration. Heterocycles 10, 425–467 (1978).
Tamura, S. et al. Isolation and physiological activities of piericidin a, a natural insecticide produced by Streptomyces. Agr. Biol. Chem. 27, 576–582 (1963).
Takahashi, N., Suzuki, A. & Tamura, S. Chemical structure of piericidin A. 2. Ozonolysis products of piericidin A derivatives. Agr. Biol. Chem. 27, 798–805 (1963).
Takahashi, N., Suzuki, A., Miyamoto, S., Tamura, S. & Mori, R. Chemical structure of piericidin A. 1. Functional groups. Agr. Biol. Chem. 27, 583–589 (1963).
Takahashi, N., Suzuki, A. & Tamura, S. Structure of piericidin A. J. Am. Chem. Soc. 87, 2066–2068 (1965).
Takahashi, N., Suzuki, A. & Tamura, S. Chemical structure of piericidin A. 3. Structures of piericidin A and octahydropiericidin A. Agr. Biol. Chem. 30, 1–12 (1966).
Suzuki, A., Takahashi, N. & Tamura, S. Chemical structure of piericidin A. 4. Structural confirmation for pyridine ring in piericidin a through synthesis. Agr. Biol. Chem. 30, 13–17 (1966).
Takahashi, N., Suzuki, A., Kimura, Y., Miyamoto, S. & Tamura, S. Structure of piericidin B and stereochemistry of piericidins. Tetrahedron Lett. 8, 1961–1964 (1967).
Takahashi, N., Yoshida, S., Suzuki, A. & Tamura, S. Chemical structure of piericidin A. 6. Stereochemistry. Agr. Biol. Chem. 32, 1108–1114 (1968).
Yoshida, S., Shiraishi, S. & Takahashi, N. Structural revision of piericidin A by combination of CMR spectroscopic and biosynthetic studies. Agr. Biol. Chem. 41, 587–591 (1977).
Yoshida, S., Shiraishi, S., Fujita, K. & Takahashi, N. Biosynthetic studies on piericidin A and its structural revision. Tetrahedron Lett. 16, 1863–1866 (1975).
Jansen, R. & Hofle, G. Revised stereochemistry of piericidin A1. Tetrahedron Lett. 24, 5485–5486 (1983).
Cox, C. M. & Whiting, D. A. Stereoselective synthesis of a synthon for the natural electron–transfer inhibitors myxalamide D and piericidin A. J. Chem. Soc. Perkin Trans. 1, 660–662 (1991).
Cox, C. M. & Whiting, D. A. Synthetic studies on electron–transport inhibitors. 1. Chiral synthesis of a synthon for myxalamide D, piericidin A, and the actinopyrones. J. Chem. Soc. Perkin Trans. 1, 1901–1905 (1991).
Yoshida, S., Yoneyama, K., Shiraishi, S., Watanabe, A. & Takahashi, N. Chemical structures of new piericidins produced by Streptomyces pactum. Agr. Biol. Chem. 41, 855–862 (1977).
Yoshida, S., Yoneyama, K., Shiraishi, S., Watanabe, A. & Takahashi, N. Isolation and physical–properties of new piericidins produced by Streptomyces pactum. Agr. Biol. Chem. 41, 849–853 (1977).
Nishioka, H. et al. Isolation and structure determination of a novel phosphatidylinositol turnover inhibitor, piericidin B1 N-oxide. J. Antibiot. 44, 1283–1285 (1991).
Nishioka, H. et al. Isolation and structure determination of novel phosphatidylinositol turnover inhibitors, piericidin B5 and B5 N-oxide, from Streptomyces sp. J. Antibiot. 46, 564–568 (1993).
Urakawa, A. et al. IT-143-A and B, novel piericidin group antibiotics produced by Streptomyces sp. J. Antibiot. 49, 1052–1055 (1996).
Ueda, J. Y. et al. A novel nuclear export inhibitor JBIR-02, a new piericidin discovered from Streptomyces sp. ML55. J. Antibiot. 60, 459–462 (2007).
Hoecker, J. & Gademann, K. Enantioselective total syntheses and absolute configuration of JBIR-02 and Mer-A2026B. Org. Lett. 15, 670–673 (2013).
Kubota, N. K. et al. Piericidins C5 and C6: new 4-pyridinol compounds produced by Streptomyces sp. and Nocardioides sp. Bioorg. Med. Chem. 11, 4569–4575 (2003).
Hayakawa, Y. et al. Piericidins C7 and C8, new cytotoxic antibiotics produced by a marine Streptomyces sp. J. Antibiot. 60, 196–200 (2007).
Hayakawa, Y. et al. Structures of new cytotoxic antibiotics, piericidins C7 and C8. J. Antibiot. 60, 201–203 (2007).
Kimura, K., Takahashi, H., Miyata, N., Yoshihama, M. & Uramoto, M. New piericidin antibiotics, 7-demethylpiericidin A1 and 7-demethyl-3'-rhamnopiericidin A1. J. Antibiot. 49, 697–699 (1996).
Chen, Y. et al. Elucidating hydroxylation and methylation steps tailoring piericidin A1 biosynthesis. Org. Lett. 16, 736–739 (2014).
Matsumoto, M. et al. New piericidin glucosides, glucopiericidins A and B. J. Antibiot. 40, 149–156 (1987).
Funayama, S. et al. Novel cytocidal antibiotics, glucopiericidinols A1 and A2. Taxonomy, fermentation, isolation, structure elucidation and biological characteristics. J. Antibiot. 42, 1734–1740 (1989).
Mori, H., Funayama, S., Sudo, Y., Komiyama, K. & Omura, S. A new antibiotic, 13-hydroxyglucopiericidin A. Isolation, structure elucidation and biological characteristics. J. Antibiot. 43, 1329–1331 (1990).
Kimura, K. et al. A new piericidin rhamnoside, 3'-rhamnopiericidin A1. J. Antibiot. 43, 1341–1343 (1990).
Iwasaki, H., Kamisango, K., Kuboniwa, H., Sasaki, H. & Matsubara, S. 3'-Deoxytalopiericidin-A1, a novel analog of antitumor antibiotics from oligotroph. J. Antibiot. 44, 451–452 (1991).
Shaaban, K. A., Helmke, E., Kelter, G., Fiebig, H. H. & Laatsch, H. Glucopiericidin C: a cytotoxic piericidin glucoside antibiotic produced by a marine Streptomyces isolate. J. Antibiot. 64, 205–209 (2011).
Schmidtchen, F. P. & Rapoport, H. Polyprenylpyridinols. Synthesis of piericidin analogues. J. Am. Chem. Soc. 99, 7014–7019 (1977).
Yoshida, S., Nagao, Y. & Takahashi, N. Synthesis of piericidin analogs, inhibitors on electron–transport system in mitochondria. Agr. Biol. Chem. 44, 2913–2920 (1980).
Yoshida, S., Nagao, Y., Watanabe, A. & Takahashi, N. Structure–activity relationship in piericidins inhibitors on the electron–transport system in mitochondria. Agr. Biol. Chem. 44, 2921–2924 (1980).
Kominato, K. et al. Mer-A2026A and B, novel piericidins with vasodilating effect. II. Physico–chemical properties and chemical structures. J. Antibiot. 48, 103–105 (1995).
Ono, M., Yoshida, N. & Akita, H. Synthetic study of piericidins. 2. Synthesis of piericidin analogues. Chem. Pharm. Bull. 45, 1745–1750 (1997).
Schnermann, M. J. et al. Total synthesis of piericidin A1 and B1 and key analogues. J. Am. Chem. Soc. 128, 11799–11807 (2006).
Schnermann, M. J. & Boger, D. L. Total synthesis of piericidin A1 and B1. J. Am. Chem. Soc. 127, 15704–15705 (2005).
Keaton, K. A. & Phillips, A. J. Titanium(II)–mediated cyclizations of (silyloxy)enynes: A total synthesis of (–)-7-demethylpiericidin A1. J. Am. Chem. Soc. 128, 408–409 (2006).
Kikuchi, R., Fujii, M. & Akita, H. Total synthesis of (+)-piericidin A1 and (–)-piericidin B1. Tetrahedron Asymmetry 20, 1975–1983 (2009).
Kikuchi, R., Fujii, M. & Akita, H. Synthetic study of piericidin A1 and B1. J. Mol. Catal. B Enzym. 62, 125 (2010).
Lipshutz, B. H. & Amorelli, B. Total synthesis of piericidin A1. Application of a modified Negishi carboalumination-nickel-catalyzed cross-coupling. J. Am. Chem. Soc. 131, 1396–1397 (2009).
Ono, M., Yoshida, N., Kokubu, Y., Sato, E. & Akita, H. Synthetic study of piericidins.1. Synthesis of the side chain of piericidin B1. Chem. Pharm. Bull. 45, 1428–1434 (1997).
Kimura, Y., Takahashi, N. & Tamura, S. Biosynthesis of piericidins A and B by Streptomyces mobaraensis. Agr. Biol. Chem. 33, 1507 (1969).
Tanabe, M. & Seto, H. Biosynthetic studies with carbon 13. Piericidin A. J. Org. Chem. 35, 2087–2088 (1970).
Liu, Q. et al. Elucidation of piericidin A1 biosynthetic locus revealed a thioesterase–dependent mechanism of alpha–pyridone ring formation. Chem. Biol. 19, 243–253 (2012).
Degli Esposti, M. et al. Complex I and complex III of mitochondria have common inhibitors acting as ubiquinone antagonists. Biochem. Biophys. Res. Commun. 190, 1090–1096 (1993).
Gutman, M., Beinert, H. & Singer, T. P. Studies on respiratory chain-linked reduced nicotinamide-adenine dinucleotide dehydrogenase. 20. Relation of respiratory chain-linked reduced nicotinamide-adenine dinucleotide dehydrogenase to energy-coupling site-1. Biochemistry 11, 556–562 (1972).
Friedrich, T. et al. Two binding sites of inhibitors in NADH: ubiquinone oxidoreductase (complex I). Relationship of one site with the ubiquinone–binding site of bacterial glucose:ubiquinone oxidoreductase. Eur. J. Biochem. 219, 691–698 (1994).
Lambert, A. J. & Brand, M. D. Inhibitors of the quinone–binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Biol. Chem. 279, 39414–39420 (2004).
Prieur, I., Lunardi, J. & Dupuis, A. Evidence for a quinone binding site close to the interface between NUOD and NUOB subunits of Complex I. Biochim. Biophys. Acta 1504, 173–178 (2001).
Gutman, M. & Kliatchko, S. Mechanism of inhibition by ubicidin – inhibitor with piericidin ring structure and ubiquinone side chain. FEBS Lett. 67, 348–353 (1976).
Degli Esposti, M., Ghelli, A., Ratta, M., Cortes, D. & Estornell, E. Natural substances (acetogenins) from the family Annonaceae are powerful inhibitors of mitochondrial NADH dehydrogenase (Complex I). Biochem. J. 301, 161–167 (1994).
Mitsui, T., Fukami, J. I., Fukunaga, K., Takahashi, N. & Tamura, S. Studies on piericidin – Antagonistic effect of vitamin-K3 on respiratory chain of insects and mammals in presence of piericidin A. Agr. Biol. Chem. 34, 1101–1109 (1970).
Chung, K., Cho, K., Asami, Y., Takahashi, N. & Yoshida, S. in Bioenergetics (eds Kim, C. & Ozawa, T.) 65–81 (Springer, 1990).
Miyoshi, H. Structure-activity relationships of some complex I inhibitors. Biochim. Biophys. Acta Bioenerg. 1364, 236–244 (1998).
Degli Esposti, M. Inhibitors of NADH–ubiquinone reductase: an overview. Biochim. Biophys. Acta 1364, 222–235 (1998).
Chung, K. H., Cho, K. Y., Asami, Y., Takahashi, N. & Yoshida, S. New 4-hydroxypyridine and 4-hydroxyquinoline derivatives as inhibitors of NADH-ubiquinone reductase in the respiratory-chain. Z. Naturforsch. C. 44, 609–616 (1989).
Horgan, D. J., Ohno, H., Singer, T. P. & Casida, J. E. Studies on respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase. 15. Interactions of piericidin with mitochondrial respiratory chain. J. Biol. Chem. 243, 5967–5976 (1968).
Takahashi, N. et al. Isolation structure and physiological activities of piericidin B nature insecticide produced by a Streptomyces. Agr. Biol. Chem. 32, 1115–1122 (1968).
Lummen, P. Complex I inhibitors as insecticides and acaricides. Biochim. Biophys. Acta 1364, 287–296 (1998).
Fato, R. et al. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta 1787, 384–392 2009.
Ohnishi, S. T. et al. A possible site of superoxide generation in the complex I segment of rat heart mitochondria. J. Bioenerg. Biomembr. 37, 1–15 (2005).
Lee, K. K. et al. Isoniazid-induced cell death is precipitated by underlying mitochondrial complex I dysfunction in mouse hepatocytes. Free Radic. Biol. Med. 65, 584–594 (2013).
Ikezawa, N., Ifuku, K., Endo, T. & Sato, F. Inhibition of photosystem II of spinach by the respiration inhibitors piericidin A and thenoyltrifluoroacetone. Biosci. Biotechnol. Biochem. 66, 1925–1929 (2002).
Hollerhage, M. et al. Piericidin A aggravates Tau pathology in P301S transgenic mice. PLoS ONE 9, e113557 (2014).
Choi, W. S., Palmiter, R. D. & Xia, Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson's disease model. J. Cell Biol. 192, 873–882 (2011).
Jeso, V., Yang, C., Cameron, M. D., Cleveland, J. L. & Micalizio, G. C. Synthesis and SAR of Lehualide B: a marine–derived natural product with potent anti–multiple myeloma activity. ACS Chem. Biol. 8, 1241–1252 (2013).
Takatsuki, A., Tamura, G. & Arima, K. Antiviral and antitumor antibiotics. XIV. Effects of ascochlorin and other respiration inhibitors on multiplication of Newcastle disease virus in cultured cells. Appl. Microbiol. 17, 825–829 (1969).
Kominato, K. et al. Mer-A2026A and B, novel piericidins with vasodilating effect. I. Producing organism, fermentation, isolation and biological properties. J. Antibiot. 48, 99–102 (1995).
Kroiss, J. et al. Symbiotic streptomycetes provide antibiotic combination prophylaxis for wasp offspring. Nat. Chem. Biol. 6, 261–263 (2010).
Sawyer, S. T. & Cohen, S. Enhancement of calcium-uptake and phosphatidylinositol turnover by epidermal growth factor in A431 cells. Biochemistry 20, 6280–6286 (1981).
Habenicht, A. J. R. et al. Early changes in phosphatidylinositol and arachidonic acid metabolism in quiescent swiss 3T3 cells stimulated to divide by platelet–derived growth factor. J. Biol. Chem. 256, 2329–2335 (1981).
Nishioka, H. et al. Isolation and structure determination of a novel phosphatidylinositol turnover inhibitor, piericidin B1 N-Oxide. J. Antibiot. 44, 1283–1285 (1991).
Nishioka, H. et al. Antitumor effect of piericidin B1 N-oxide. J. Antibiot. 47, 447–452 (1994).
Ahn, S. C. et al. Inhibition of PDGF–induced phosphoinositide–turnover by glucopiericidin A. Biochem. Mol. Biol. Int. 37, 125–132 (1995).
Debbas, M. & White, E. Wild-type P53 mediates apoptosis by E1a, which is inhibited by E1b. Gene Dev. 7, 546–554 (1993).
Kitagawa, M., Ikeda, S., Tashiro, E., Soga, T. & Imoto, M. Metabolomic identification of the target of the filopodia protrusion inhibitor glucopiericidin A. Chem. Biol. 17, 989–998 (2010).
Hwang, J. H. et al. Etoposide-resistant HT-29 human colon carcinoma cells during glucose deprivation are sensitive to piericidin A, a GRP78 down–regulator. J. Cell Physiol. 215, 243–250 (2008).
Kuo, J. F., Dill, I. K. & Holmlund, C. E. Effects of piericidin a on metabolism of isolated adipose cells. Biochem. Pharmacol. 17, 867–872 (1968).
Magae, J. et al. Screening for specific inhibitors of phagocytosis of thioglycollate-elicited macrophages. Biosci. Biotechnol. Biochem. 58, 104–107 (1994).
Ahn, S. C. et al. Screening of interleukin–2 production inhibitor with mouse thymoma EL4 cells. J. Antibiot. 55, 1013–1015 (2002).
Duncan, M. C. et al. An NF-κB-based high-throughput screen identifies piericidins as inhibitors of the Yersinia pseudotuberculosis type III secretion system. Antimicrob. Agents Chemother. 58, 1118–1126 (2014).
Acknowledgements
This work is a result of financial support from the National Institutes of Health, National Cancer Institute under grant CA044848 (to WF) and from the China Scholarship Council (to XZ). This article is dedicated to the fond memory of the late professor Lester Mitscher, a great scholar, teacher and emeritus editor of this Journal.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Zhou, X., Fenical, W. The unique chemistry and biology of the piericidins. J Antibiot 69, 582–593 (2016). https://doi.org/10.1038/ja.2016.71
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2016.71
This article is cited by
-
Genome-based discovery and total synthesis of janustatins, potent cytotoxins from a plant-associated bacterium
Nature Chemistry (2022)
-
The immunotoxicity of ten insecticides against insect hemocyte cells in vitro
In Vitro Cellular & Developmental Biology - Animal (2022)
-
Special issue dedicated to William Fenical: a pioneer in marine/marine-derived microbial chemistry
The Journal of Antibiotics (2020)
-
Glycosylated piericidins from an endophytic streptomyces with cytotoxicity and antimicrobial activity
The Journal of Antibiotics (2018)
