
Abstract
Background
Pediatric clinical trials commonly experience recruitment challenges including limited number of patients and investigators, inclusion/exclusion criteria that further reduce the patient pool, and a competitive research landscape created by pediatric regulatory commitments. To overcome these challenges, innovative approaches are needed.
Methods
This article explores the use of Bayesian statistics to improve pediatric trial feasibility, using pediatric Type-2 diabetes as an example. Data for six therapies approved for adults were used to perform simulations to determine the impact on pediatric trial size.
Results
When the number of adult patients contributing to the simulation was assumed to be the same as the number of patients to be enrolled in the pediatric trial, the pediatric trial size was reduced by 75–78% when compared with a frequentist statistical approach, but was associated with a 34–45% false-positive rate. In subsequent simulations, greater control was exerted over the false-positive rate by decreasing the contribution of the adult data. A 30–33% reduction in trial size was achieved when false-positives were held to less than 10%.
Conclusion
Reducing the trial size through the use of Bayesian statistics would facilitate completion of pediatric trials, enabling drugs to be labeled appropriately for children.
Similar content being viewed by others

Main
The field of pediatric research has experienced impressive growth as a result of the United States (US) and the European Union regulations that require newly developed drugs to be studied in children (1). Pediatric studies are typically initiated after approval of the adult indication, which allows a positive benefit/risk assessment to be first established in adults. During execution of the adult clinical program, the US Food and Drug Administration (FDA) and European Medicines Agency require and approve plans for the pediatric trials, known as Pediatric Study Plans in the US and Pediatric Investigation Plans (PIPs) in Europe. However, execution of the pediatric trials is often more difficult than the adult trials because of there being generally far fewer pediatric patients and the complexities of studying a vulnerable patient population. In addition, the regulations that have spurred the research by requiring trials for each newly developed drug have also resulted in a competitive pediatric clinical trial landscape for certain indications. The challenges encountered in conducting pediatric trials necessitate innovative approaches to the design and conduct of these trials. In this paper we explore the approach of utilizing Bayesian statistics in simulating pediatric clinical trials of Type-2 diabetes (T2D) drugs as an illustrative example.
Pediatric T2D trials exemplify the challenges encountered in pediatric research. In addition to there being an insufficient pediatric trial infrastructure and inclusion/exclusion criteria that reduce the available patient pool, T2D trials also contend with a limited number of patients, whose demographic characteristics further impair recruitment, and a competitive research landscape. Currently in the US and European Union, only metformin and insulin are approved for pediatric use. A plethora of agents have been approved or are in development for treatment of adult patients, and, for recently developed drugs, pediatric development plans are in place. As of early 2017, PIPs had been approved for 24 products (nine glucagon-like peptide-1 (GLP-1) agonists, five dipeptidyl peptidase-4 inhibitors, six sodium-glucose co-transporter 2 inhibitors, one sodium-glucose co-transporter 1/2 inhibitors, one G-protein-coupled receptor 40 agonist, one GLP-1 and glucagon receptor co-agonist, and one dopamine agonist) that are being developed by 15 companies. Of these products, 12 are already marketed for use in adults in the European Union. The products will also need to have approved Pediatric Study Plans in the US, but because US pediatric commitments are not publicly disclosed until product approval for the adult indication, PIPs provide a more complete accounting of patient recruitment needs. Using an average of 224 patients per T2D PIP (2), over 5,000 patients would be needed to satisfy current PIP commitments, with half of those patients needed now to support trials for products that have already been approved in adults.
On the basis of the data from the SEARCH for Diabetes in Youth Study, the prevalence of T2D in children of 10–19 years of age was estimated to be 0.46 per 1,000 children in 2009, or roughly 20,000 cases (3, 4). Assuming a projected yearly increase of 2.3% (ref. 5), fewer than 25,000 pediatric patients diagnosed with T2D are estimated to currently be in the US, and of those patients, only 500–600 are estimated to be eligible trial subjects, for reasons elaborated below (6). Prevalence in European countries is far less than that in the US, offering a minimal increase in the patient pool for clinical trials. Of the limited number of pediatric patients diagnosed with T2D, only a small percentage qualifies for inclusion in clinical trials. Metformin, which is recommended by the American Academy of Pediatrics as first-line therapy (7), provides adequate control for many patients (up to 50% of patients in the Treatment Options for Type 2 Diabetes in Adolescents and Youth (TODAY) study) (8), sizably diminishing the patient pool. In addition, insulin use has traditionally been an exclusion criteria, which eliminates approximately half of the pediatric patient population, although recently this exclusion criterion has been removed from some trials (9). Additional inclusion/exclusion criteria (e.g., required hemoglobinA1c range, major medical conditions, concomitant meds, and prior diabetes medication use) further shrink the available patient pool.
In addition to having only a small patient pool, patient demographics contribute to recruitment challenges. The vast majority of pediatric patients are minorities, with most coming from socioeconomically disadvantaged environments as evidenced by annual family income or dependence on government insurance (10, 11). In the US, the higher rates of uninsured and the associated decreased access to healthcare experienced by minorities make it less likely that these patients will be aware of or participate in clinical trials (12). Patients from lower socioeconomic environments also encounter logistical impediments to enrolling in and completing clinical trials such as address changes, limited transportation options, and the inability of parents to miss work in order to bring the child to study visits (9, 13).
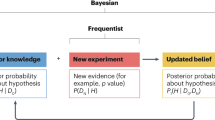
The difficulties described above in recruiting patients into pediatric T2D trials are applicable in other challenging pediatric indications and have led to proposals for innovative approaches that include improving trial infrastructure, broadening inclusion/exclusion criteria, and using novel study designs (9, 13). In this paper, we conduct an elementary exploration of the application of Bayesian statistics to pediatric T2D trials to illustrate the potential of this approach to enhance the feasibility of pediatric trials. Bayesian statistics incorporates prior knowledge or beliefs about the effect of a treatment into the final conclusions of a study. This is accomplished by using an assumed distribution for the model parameters and then merging them with the distribution estimated from the data collected in the study to form the posterior distribution. Subsequent inferences about the treatment effect are based on the posterior distribution, which in this case is a weighted average of knowledge gained from the pediatric trial and the pre-existing adult information. The inclusion of prior information allows for a more precise conclusion; therefore, the sample size of the trial can be reduced, making the trial more feasible. For those interested in advanced Bayesian methodologies, the Bayesian Statistics Working Group of the Drug Information Association recently published a paper examining the application of a variety of Bayesian methodologies to pediatric trials (14).
The impact of Bayesian methods on sample size for adult studies has been investigated in a variety of settings (15, 16). To warrant using a Bayesian approach for pediatric trials, it must be reasonable to assume that the pathophysiology of the disease, which has an impact on the relevance of the drug mechanism of action, and the absorption, distribution, metabolism, and excretion (ADME) of the drug are similar in adults and the pediatric age group being studied. The latter determination should take into account maturation of systems involved in absorption and excretion (e.g., renal, hepatic, and gastric), as well as the ontogeny of relevant metabolic enzymes and drug transporters. Finally, the dose–exposure relationship in children must be known, or plans to confirm it should be incorporated into the pediatric trial.
Although the concept of applying Bayesian statistics to pediatric trials has been discussed by experts in academia, pharmaceutical companies, and regulatory agencies (5, 14, 17, 18), little has been published that quantifies the impact of Bayesian assumptions on the sizing of pediatric trials. In this paper we utilized data from pivotal T2D adult studies conducted with six drugs, two from each of the three recently approved drug classes, to create informative priors for the treatment effect on hemoglobinA1c (HbA1c). We then ran multiple simulations for each agent in which we varied the weight given to the adult prior information in order to determine the impact on both the pediatric trial sample size and Type-I error, often referred to as the false-positive rate. The results demonstrate the potential for the use of Bayesian statistics to facilitate the completion of pediatric studies.
Methods
We utilized knowledge about the effectiveness of six drugs approved for treatment of adults with T2D to create informative priors for HbA1c treatment effect parameters. The drugs are: canagliflozin (Janssen Pharmaceuticals, Titusville, NJ) and dapagliflozin (AstraZeneca Pharmaceuticals, Wilmington, DE)—both SGLT-2 inhibitors; sitagliptin (Merck, Whitehouse Station, NJ) and linagliptin (Boehringer Ingelheim International GmbH, Ingelheim, Germany)—both DPP-4 inhibitors; and liraglutide (Novo Nordisk A/S, Bagsvaerd, Denmark) and dulaglutide (Eli Lilly and Company, Indianapolis, IN)—both GLP-1 agonists. The adult T2D clinical trial data used for creation of the prior distributions are summarized in Table 1. The trials were selected in which the drug being investigated was added to metformin therapy and the effect on HbA1c level was compared with the effect of metformin alone at 24–26 weeks. We selected data from these trials because they are likely to be the most relevant to pediatric trials, considering American Academy of Pediatrics’s recommendation that metformin be first-line therapy. For drugs that have more than one dose approved, data for the lowest approved dose were used to develop the prior distribution.
We ran simulations to investigate the power to detect a treatment difference (investigational drug+metformin vs. metformin) in which we varied the weight of the contribution of the adult prior relative to the to-be-conducted pediatric clinical trial. Ten thousand repetitions were used for each simulation, using the open source programming language of R. The posterior distribution, which can be thought of as a weighted average from the pediatric trial and the pre-existing adult information, was then derived assuming a normal distribution for both adult and pediatric patients. We further assumed the mean treatment-effect size and SD to be the same in adults and children because the fundamental pathophysiology of the disease (19, 20), the mechanism of action of the drugs, and the relevance of the HbA1C endpoint do not differ between adolescents and adults. Supporting this assumption, a similar treatment effect was achieved in adults and children treated with metformin, the only drug for which pivotal data are available in both patient populations (Table 2) (21, 22). Should data for a drug warrant assuming a different treatment effect size or SD in children, different assumptions can be incorporated into the simulation. When planning a study, a robustness or sensitivity analysis should be performed to understand the effects of mis-specification.
For each drug, the prior distribution for the treatment effect was calculated using the estimated mean and SD from the adult study. A prior for the treatment difference was used rather than separate priors for the treatment and control groups, as this was the information available for each of the drugs we evaluated. Because of the comparatively large adult sample size, using the entire sample size to calculate the prior would result in the adult study strongly influencing the analysis of the pediatric study. To reduce the influence of the prior distribution, we used an artificially smaller sample size that was fixed as a ratio to the sample size for the planned pediatric study. To simulate data for the pediatric patients, data were randomly generated from a normal distribution using the mean and SD of the assumed adult population parameters for the treatment difference relative to control.
For the initial simulations, we equally weighted the adult data to the pediatric data that will be collected in the to-be-performed trial. This means that the prior distribution for the treatment effect was calculated using the estimated mean and SD from the adult study and the sample size of the pediatric study. However, when the study was powered at 90%, an equally weighted prior still had a large influence on the analysis of the pediatric study. Therefore, in subsequent simulations we decreased the ratio of the number of adult patients to pediatric patients in an iterative manner to achieve a Type-1 error of <10% while maintaining at least 90% power. The value of 10% was arbitrarily chosen, and selection of an appropriate value is a topic considered in the Discussion.
For each of the 10,000 repetitions simulated per case, after finding the posterior distribution using the historical prior and the simulated pediatric data, the particular repetition was considered a success if 97.5% of the posterior distribution was above the value of 0, implying a treatment benefit. The minimum probability that justifies use of a treatment is debatable and may depend partly on the importance of the potential clinical benefit relative to risk. The 97.5% value was chosen to align with the typical frequentist Type-I error. If the simulated pediatric data were generated from a distribution with no treatment difference using the same prior distribution, then those cases that also satisfied this condition were considered to be a Type-I error.
Results
For all drugs that we evaluated, the initial Bayesian simulation that used an equal weighting of adult prior and pediatric trial data resulted in an estimated pediatric sample size that was 75–78% smaller than the sample size estimated using frequentist statistics, while achieving 90% power (Table 3). However, these notable decreases in sample size were associated with Type-1 errors ranging from 34 to 45%. Results were consistent for both members of each drug class, and across all three drug classes.
In subsequent simulations the weight of the adult prior to pediatric trial data was decreased to exert greater control over Type-1 error. Table 4 presents the parameters necessary for the simulations to result in a Type-1 error of less than 10% while still achieving 90% power to detect a treatment effect. The weightings varied from 1:5 for linagliptin to 1:7 for dulaglutide, with canagliflozin, dapagliflozin, sitagliptin, and liraglutide all requiring 1:6 weightings. Because dulaglutide had a small sample size estimated by frequentist statistics, changes of one subject per group had a great impact on the simulation parameters. Because the weight of the adult prior is directly related to the reduction in pediatric trial sample size, the reduction in sample size was less than that in the initial simulations and ranged from 30 to 33%.
The simulations summarized in Table 5 illustrate that accepting a slightly greater Type 1 error of 13–14% allows the pediatric trial sample size to be decreased by 40–44%. For all six drugs evaluated, a 1:4 weighting of the adult prior to pediatric trial data produced this reduction in sample size. These simulations maintained approximately half of the reduction in sample size achieved in the 1:1 weighted simulations, while reducing the Type-1 error by threefold.
The results of the simulations described in Tables 3, 4, 5 depict how applying Bayesian statistics in the context of T2D could be used to significantly reduce the sample size required for pediatric clinical trials. If the assumption that the effect size is the same in adults and in children is incorrect, and the true effect size is less in children, then the power to detect the treatment effect will be decreased. To explore this, we ran simulations for all drugs in which the actual effect size in children is half of the effect size in adults. Under these conditions the original 90% power was reduced to 69–74%, 53–58%, and 48–56% for the 1:1, 1:4, and 1:5–7 simulations, respectively. Even though the power is diminished, the power remains greater than that with the frequentist approach. In the case where there is no treatment effect, the probability of erroneously concluding that there is a treatment benefit is reflected in the Type-1 error. The Type-1 error will be greater than the typical one-sided α=2.5% associated with a frequentist statistical approach because of the assumption of benefit that is inherent in the Bayesian approach; however, the simulations illustrate how this can be limited by decreasing the weight of the adult prior.
Discussion
Regulations have resulted in an increase in pediatric research, with the goal of including information in drug labeling to guide physicians in providing appropriate care to pediatric patients. However, pediatric trials for certain indications have proven notoriously difficult to enroll, as exemplified by pediatric T2D trials, necessitating consideration of innovative approaches. Summarizing this predicament, one key opinion leader has stated that “There are simply too few eligible study subjects to be recruited by too many competing studies.” (13).
To improve the feasibility of pediatric clinical trials, we explored the use of Bayesian statistics as an alternative to the conventional statistical approach. FDA recently identified pediatric diseases that have adult trial data available as ripe for the application of Bayesian methods, which offer a means to provide comparable information in a more timely manner with fewer patients than are required by frequentist statistics (18). T2D presents an ideal indication to explore this approach, with numerous therapeutics from multiple drug classes recently approved for treatment of adults, but with only metformin and insulin approved for the treatment of children. The T2D indication is particularly apt for the investigation of Bayesian statistics because it affects adolescents, but not young children for whom justification and weighting of the adult prior would be more complex. The clinical trial simulations we performed for six antihyperglycemic agents from three different drug classes demonstrate the value that a Bayesian statistical approach could bring to pediatric trials. We acknowledge that such approaches do not address whether the sample size required to demonstrate efficacy also results in a sufficient safety database. However, alternatives to placebo-controlled trials exist for the collection of additional safety data and in the absence of a safety signal, all data need not necessarily be collected before licensure.
In order to utilize data from adult trials as informative prior data for pediatric trials, it must first be determined whether it is reasonable to assume that the adult data are relevant to the pediatric patient population. For T2D therapies, which we used to explore the Bayesian statistical approach, this is a reasonable assumption because the pediatric patients are adolescents, with similar body weights and body mass indices as adults, and the underlying insulin resistance and progressive β-cell deterioration make the therapeutic mechanisms of action equally relevant in both populations (19, 20). The supposition that adolescents and adults exhibit similar ADME of T2D medicines is confirmed by data showing comparable pharmacokinetic parameters in both groups (9, 23, 24, 25, 26). It is acknowledged, however, that β-cell decline occurs more rapidly in children, and hormonal changes associated with puberty may exacerbate insulin resistance (6, 27, 28). For these reasons, whereas the SD observed in the adult trial was utilized for the pediatric trial simulations we performed, it may be worthwhile to consider increasing the SD assumed for pediatric subjects so as to be more conservative in the assumptions. In the absence of pediatric data to guide the determination of the SD, another possibility may be to adjust the SD assumption based on a blinded interim assessment made during the pediatric trial.
Notwithstanding the justification provided above for the utilization of adult prior data, no exploration of the application of Bayesian statistics to pediatric T2D trials would be complete without acknowledging that pediatric trials of some T2D drugs have failed to meet pre-specified efficacy criteria. However, it would be incorrect to conclude on the basis of these trials that the drugs do not work in children. A closer examination of the pediatric and adult trials reveals significant differences in design between them that likely explain the seemingly disparate results. Furthermore, for each drug tested there is evidence of a treatment effect in children, either within the subset of treatment-naive subjects, for whom there is not the substantial confounder of pre-trial therapy, and/or from a subsequent, better-designed trial. Given the importance to the Bayesian approach of the assumption that the adult data are relevant for pediatric trials, additional details regarding why some pediatric T2D trials failed to definitively demonstrate efficacy are provided below. The totality of the evidence is consistent with the expectation that children respond to T2D treatments in generally the same manner as adults, and it supports the use of informative priors derived from the adult trials for the application of Bayesian statistics to pediatric trials.
Christensen et al. (23) recently reviewed the pediatric drug development programs for non-insulin T2D therapies that have been reviewed by FDA, namely glimepiride, rosiglitazone, glyburide/metformin, and metformin (23). Only the metformin program resulted in a labeled pediatric indication; the complexities and weaknesses encountered with the other programs are summarized here. In contrast to the adult trials, the pediatric trials for glimepiride and rosiglitazone were conducted as non-inferiority (NI) trials, a design that is complicated by issues of assay sensitivity, selection of the NI margin, and large required sample size. Both trials appear underpowered for this analysis, which likely explains why NI was not demonstrated (26, 29). FDA’s review of glimiperide indicates that the SD had been assumed to be 1.2%, but proved to be ~2.0% in the trial, which resulted in a power of only 40%. In addition, a significant confounder in the trials for rosiglitazone, glimperide, and glyburide/metformin was the inclusion of non-naive patients.
The trial of glyburide/metformin was not a NI trial (it was a superiority trial comparing the combination with the individual components); however, demonstration of a treatment effect was significantly hampered in this trial, as well as the trials of rosiglitazone and glimiperide, by inclusion of non-naive patients. The duration of washout of prior therapy was short or nonexistent in these trials for ethical reasons; therefore, the HbA1c values measured at randomization had not returned to pretreatment values (26, 30, 31). Because non-naive patients accounted for nearly half the patients enrolled in each of the trials, this trial design would be expected to noticeably hinder the ability to demonstrate an impact on HbA1c by the drug being tested. There was, however, evidence of an impact of treatment in the naive population. Reductions in HbA1c from baseline (mean change±SEM) for naive and non-naive patients were −1.35±2.00 and −0.09±1.63, respectively, for glyburide/metformin, and −0.97±0.3 and +0.17±0.7, respectively, for glimepiride (29, 31). Although it is not stated specifically whether the effects on HbA1c in naive subjects were statistically significant, presumably this is the case for glimepiride because it was able to produce a statistically significant decrease in the combined (naive plus non-naive) population. On the basis of this result, the FDA reviewer concluded that “Glimepiride and metformin were both effective in achieving glycemic control from baseline to endpoint in pediatric subjects with Type-2 diabetes mellitus.” (29) Nevertheless, the trial was considered to have failed because the primary endpoint of NI to metformin was not demonstrated. Of note, the glimepiride trial utilized the most stringent HbA1c entry criteria of the three trials, >7.1%, which may also have contributed to the ability to demonstrate a treatment effect. For glyburide/metformin, the FDA reviewer posits that having very few patients in the pediatric trial with baseline HbA1c >9% accounts for the inability to detect a treatment effect because it was patients with this level of hyperglycemia that drove the treatment effect in the adult trial (32).
Data for non-naive patients are not available for rosiglitazone, but the impact of prior therapy can still be seen by comparing the effect achieved in naive patients with that in all-randomized patients, −0.32±1.64 and −0.14±1.52 (mean change±SD), respectively (26). For this latter comparison the P values were 0.1552 and 0.3629, respectively, and, although statistical significance was not achieved, the lower P value for naive patients was achieved despite the smaller number of patients in the naive subgroup (55 vs. 97). It is also noteworthy that when rosiglitazone was studied in a subsequent trial of different design, it was shown to be effective in maintaining glycemic control when added to metformin therapy (8).
For the three drug products described above, the weight of evidence suggests that they are effective in children. An evaluation of efficacy, however, is not sufficient to conclude that drugs should be recommended treatments for children. To make an approvability decision, a risk-benefit assessment is also needed, which would account for any negative consequences of treatment. For the drugs described above, weight gain is a known treatment-related effect, and FDA cited this as a factor in its decisions to not grant pediatric indications for the drugs.
Having expounded on the rationale for using a Bayesian approach that applies informative priors derived from adult trials to pediatric trials, selection of the extent to which the prior contributes to the posterior distribution (i.e., the weight given to the prior) remains to be discussed. One of the principles of Bayesian statistics is that the weight given to the prior should correlate with the confidence in the relevance of the prior to the new study. For our simulations we initially used a weight of 1:1 in order to provide a point of reference, i.e., we assumed the number of adult patients contributing to the posterior data set was the same as the number of pediatric patients to be enrolled in the trial. However, results from the 1:4 to 1:7 simulations provided a better balance between the goals of reducing the trial sample size and controlling the Type-1 error. Given that we used T2D drugs for our simulations, selection of a weight in this range seemed reasonable in light of the discussion above regarding the evidence for similar effectiveness of T2D treatments in adults and children. We would suggest that the 1:4 weighting provides the most appropriate balance, maintaining approximately half of the reduction in sample size that was achieved in the 1:1 weighted simulation, while reducing the Type-1 error by threefold. It is noted that the Type-1 error would be increased if the SD in children is greater than that in the adult prior.
There have been other Bayesian methodologies developed, such as the use of a normalized power prior (33, 34), which also effectively control the influence of the assumed prior on the final analysis. This is accomplished by reducing the magnitude of the prior function in the calculation of the posterior distribution. Instead, we have adjusted the parameters of the prior directly to control the prior’s influence.
The results of our simulations, using T2D drugs as examples, demonstrate that applying Bayesian statistics to pediatric trials offers a means of facilitating trial completion by reducing trial size. Inherent in the methodology is an assumption of benefit that results in Type-1 error values greater than those used in frequentist statistics, but this is because the calculation of Type-1 error presumes that the assumption of benefit is incorrect. If the assumption of benefit is justified, then the description of Type-1 error as the probability of a false-positive is misleading. Furthermore, the assumption of benefit can be tempered by limiting the weight of the adult prior. Our exploration of the application of Bayesian methodology to pediatric research leads us to conclude that this approach should be further investigated as a viable means to enhance the feasibility of completing pediatric trials that are needed to support labeling of drugs for pediatric use.
References
Turner MA, Catapano M, Hirschfeld S et al. Paediatric drug development: the impact of evolving regulations. Adv Drug Delivery Rev 2014;73:2–13.
Karres J, Tomasi P . New medicines for type 2 diabetes in adolescents: many products, few patients. Expert Rev Clin Pharmacol 2013;6:227–9.
Dabelea D, Mayer-Davis EJ, Saydah S et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA 2014;311:1778–86.
Pettitt DJ, Talton J, Dabelea D et al. Prevalence of diabetes in U.S. youth in 2009: the SEARCH for diabetes in youth study. Diabetes Care 2014;37:402–8.
European Medicines Agency Report of the workshop on paediatric investigation plans in type 2 diabetes mellitus. EMA/272764/2013, 2013 (http://www.ema.europa.eu/docs/en_GB/document_library/Report/2013/05/WC500143022.pdf) Accessed 1 August 2016.
Nadeau KJ, Anderson BJ, Berg EG et al. Youth-onset type 2 diabetes consensus report: current status, challenges, and priorities. Diabetes Care 2016;39:1635–42.
Copeland KC, Silverstein J, Moore KR et al. Management of newly diagnosed type 2 diabetes mellitus (T2DM) in children and adolescents. Pediatrics 2013;131:364–82.
Zeitler P, Hirst K, Pyle L et al. A clinical trial to maintain glycemic control in youth with type 2 diabetes. N Engl J Med 2012;366:2247–56.
Tamborlane WV, Haymond MW, Dunger D et al. Expanding treatment options for youth with type 2 diabetes: current problems and proposed solutions: a white paper from the NICHD Diabetes Working Group. Diabetes Care 2016;39:323–9.
Copeland KC, Zeitler P, Geffner M et al. Characteristics of adolescents and youth with recent-onset type 2 diabetes: the TODAY cohort at baseline. J Clin Endocrinol Metab 2011;96:159–67.
Nambam B, Silverstein J, Cheng P et al. A cross-sectional view of the current state of treatment of youth with type 2 diabetes in the USA: enrollment data from the Pediatric Diabetes Consortium Type 2 Diabetes Registry. Pediatr Diabetes 2016;18:222–9.
Ortiz B and Guerrero S. Recruitment and retention of minority populations in clinical trials. In: Mulberg AE, Murphy D, Dunne J, and Mathis L, eds. Pediatric Drug Development: Concepts and Applications, 2nd edn, John Wiley & Sons Ltd: Chichester, UK, 2013:472-476.
Zeitler P, Chou HS, Copeland KC et al. Clinical trials in youth-onset type 2 diabetes: needs, barriers, and options. Curr Diab Rep 2015;15:28.
Gamalo-Siebers M, Savic J, Basu C et al, Statistical modeling for Bayesian extrapolation of adult clinical trial information in pediatric drug evaluation, 2016 (http://www.bayesianscientific.org/wp-content/uploads/2016/11/Pediatric_BSWG_Aug28_2016.pdf) Accessed 11 April 2017.
Combest AJ, Wang S, Healey BT et al. Alternative statistical strategies for biosimilar drug development. GaBI J 2014;3:13–20.
Wandel S, Roychoudhury S . Designing and analysing clinical trials in mental health: an evidence synthesis approach. Evid Based Mental Health 2016;19:114–7.
Schoenfeld DA, Zheng H, Finkelstein DM . Bayesian design using adult data to augment pediatric trials. Clin Trials 2009;6:297–304.
Lavange L. American course on drug development and regulatory sciences special workshop: substantial evidence in 21st century regulatory science. CDER and CBER Experience, 2016 (http://pharmacy.ucsf.edu/events/2016/04/evidence) Accessed 1 August 2016.
Giannini C, Weiss R, Cali A et al. Evidence for early defects in insulin sensitivity and secretion before the onset of glucose dysregulation in obese youths: a longitudinal study. Diabetes 2012;61:606–14.
Weiss R, Caprio S, Trombetta M et al. Beta-cell function across the spectrum of glucose tolerance in obese youth. Diabetes 2005;54:1735–43.
DeFronzo RA, Goodman AM . Efficacy of metformin in patients with non-insulin-dependent diabetes mellitus. The Multicenter Metformin Study Group. N Engl J Med 1995;333:541–9.
Jones KL, Arslanian S, Peterokova VA et al. Effect of metformin in pediatric patients with type 2 diabetes: a randomized controlled trial. Diabetes Care 2002;25:89–94.
Christensen ML, Franklin BE, Momper JD et al. Pediatric drug development programs for type 2 diabetes: a review. J Clin Pharmacol 2015;55:731–8.
Gao X, Christensen M, Burghen GA et al. Pharmacokinetics of metformin in pediatric type 2 diabetic and healthy adult subjects. Clin Pharmacol Ther 2003;73:P46.
Gottschalk M, Danne T, Vlajnic A et al. Glimepiride versus metformin as monotherapy in pediatric patients with type 2 diabetes: a randomized, single-blind comparative study. Diabetes Care 2007;30:790–4.
FDA Clinical Review of NDA 21-071S-015, 2005 (http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM428704.pdf) Accessed 2 August 2016.
Rosenbloom AL, Silverstein JH, Amemiya S et al. Type 2 diabetes in children and adolescents. Pediatr Diabetes 2009;10 (Suppl 12): 17–32.
Amiel SA, Sherwin RS, Simonson DC et al. Impaired insulin action in puberty. A contributing factor to poor glycemic control in adolescents with diabetes. N Engl J Med 1986;315:215–9.
FDA statistical review and evaluation of NDA 20-496/SE5-015, 2005 (http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM376919.pdf) Accessed 1 August 2016.
FDA Clinical Review of NDA 20-496/SE5-015, 2005 (http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM376893.pdf) accessed 01 August 2016.
FDA statistical review and evaluation of NDA 21-178/-007, 2003 (http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM445509.pdf) Accessed 1 August 2016.
FDA clinical review of NDA 21-178 (http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM445508.pdf) Accessed 04 August 2016.
Ibrahim JG, Chen M-H . Power prior distributions for regression models. Stat Sci 2000;15:46–60.
Duan Y, Ye K, Smith EP . Evaluating water quality using power priors to incorporate historical information. Environmetrics 2006;17:95–106.
Lavalle-Gonzalez FJ, Januszewicz A, Davidson J et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomised trial. Diabetologia 2013;56:2582–92.
Bailey CJ, Gross JL, Hennicken D et al. Dapagliflozin add-on to metformin in type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled 102-week trial. BMC Med 2013;11:43–52.
Taskinen MR, Rosenstock J, Tamminen I et al. Safety and efficacy of linagliptin as add-on therapy to metformin in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled study. Diabetes Obes Metab 2011;13:65–74.
Charbonnel B, Karasik A, Liu J et al. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006;29:2638–43.
Nauck M, Weinstock RS, Umpierrez GE et al. Efficacy and safety of dulaglutide versus sitagliptin after 52 weeks in type 2 diabetes in a randomized controlled trial (AWARD-5). Diabetes Care 2014;37:2149–58.
Nauck M, Frid A, Hermansen K et al. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care 2009;32:84–90.
Acknowledgements
We thank J.D., Corporate Communications Consultant to QuintilesIMS, for her editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
During the completion of this research, all authors were employees of Quintiles (which became QuintilesIMS), a company that provides clinical trial services to biopharmaceutical companies.
Additional information
STATEMENT OF FINANCIAL SUPPORT
This research received no financial support from extramural sources.
Rights and permissions
About this article

Cite this article
Huff, R., Maca, J., Puri, M. et al. Enhancing pediatric clinical trial feasibility through the use of Bayesian statistics. Pediatr Res 82, 814–821 (2017). https://doi.org/10.1038/pr.2017.163
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2017.163
This article is cited by
-
The Next Horizon of Drug Development: External Control Arms and Innovative Tools to Enrich Clinical Trial Data
Therapeutic Innovation & Regulatory Science (2024)
-
Randomized Controlled Trials in Pediatric Cardiology: A Power Struggle?
Pediatric Cardiology (2023)
-
Bayesian adaptive design for pediatric clinical trials incorporating a community of prior beliefs
BMC Medical Research Methodology (2022)
