
Abstract
Liver receptor homolog-1 (LRH-1), a member of the nuclear receptor superfamily, is a ligand-regulated transcription factor that plays crucial roles in metabolism, development, and immunity. Despite being classified as an ‘orphan’ receptor due to the ongoing debate surrounding its endogenous ligands, recent researches have demonstrated that LRH-1 can be modulated by various synthetic ligands. This highlights the potential of LRH-1 as an attractive drug target for the treatment of inflammation, metabolic disorders, and cancer. In this review, we provide an overview of the structural basis, functional activities, associated diseases, and advancements in therapeutic ligand research targeting LRH-1.
Similar content being viewed by others


Introduction
Nuclear receptors (NRs) are a class of ligand-dependent transcriptional factors that directly bind to DNA to regulate the expression of the target gene, typically in the presence of specific ligands such as steroids, thyroid hormones, lipophilic vitamins and cholesterol metabolites [1]. The human NR superfamily consists of 48 members, classified into 7 subfamilies (NR0-NR6) based on their sequence homology.
Structurally, NRs possess modular and conserved domains with highly consistent sequences, consisting of four major functional regions. The A/B region is the N-terminal regulatory domain that contains the ligand-independent activation function domain (AF-1) [2]. The C region is a highly conserved DNA-binding domain (DBD) that contains two zinc finger motifs, enabling binding to specific DNA sequences known as hormone response elements (HRE) [3]. The D region is a flexible hinge region that bridges the DBD and the ligand binding domain (LBD). The E region is the C-terminal region that contains the ligand-binding domain (LBD) and the ligand-dependent activation function domain (AF-2). The AF-1 and hinge regions exhibit the greatest diversity, while the DBD and LBD regions show the highest conservation in sequence and structure across the superfamily. When a ligand binds to the LBD, it induces a conformational change of the binding pocket, thus altering the orientation of AF-2, driving the recruitment of cofactors. Subsequently, the resulting complex translocates to the nucleus, where it binds to specific DNA-recognized binding sites, thereby affecting the expression of downstream genes [4].
Functionally, NRs play critical roles in a wide range of physiological processes, such as development, metabolism, proliferation and homeostasis [5]. Dysregulation of NR signaling is associated with diverse metabolic diseases, inflammations, autoimmune disorders and cancer. Therefore, the NR superfamily has emerged as the main therapeutic targets for human diseases. NRs modulators, such as tamoxifen, dexamethasone, rosiglitazone, and enzalutamide, account for 13% in all FDA-approved drugs [6].
Liver receptor homolog-1 (LRH-1; NR5A2), classified into the Ftz-F1 subfamily of the nuclear receptors, is considered as orphan nuclear receptor due to the lack of known endogenous ligands [7]. Over the past decade, extensive research has revealed that LRH-1 is involved in a variety of physiological and pathological processes in human, thereby highlighting its therapeutic potential for a wide range of diseases, such as metabolic disorders, inflammation and cancer [8,9,10,11,12,13].
In this review, we will explore the structural basis of LRH-1 function, its normal physiological function, as well as related diseases. Additionally, we will provide an overview of the current status of LRH-1 ligands research and development.
Structural basis and functions
LRH-1 shares the typical modular structure of NRs with four functional domains labeled A to E, belonging to the NR5 subfamily along with the highly homologous steroidogenic factor 1 (SF-1, Fig. 1a, b) [7, 14]. It has been reported that the NR5 subfamily members have certain unique structural domains compared to most nuclear receptors, which contributes to some distinct functional features.

a Schematic diagram of the domain structure of NR5 subfamily members. LRH-1 and SF-1 display conserved modular domain architecture with an N-terminal domain lacking activation function 1 (AF-1), followed by a DNA binding domain (DBD) containing two extra structures named C-terminal extension (CTE) and Ftz-F1 helix, a hinge domain and an LBD with an activation function 2 (AF-2) domain. b Sequence alignment of the LBD of hLRH-1 and human SF-1 (hSF-1). A cartoon presentation of the overall architecture of hLRH-1 is depicted below the sequence alignment. Identical residues are labeled with an asterisk. Partially conserved residues are labeled with a colon. The residue numbering for hLRH-1 and hSF-1 are S300-A541 and N222-T461, respectively. Residues of hLRH-1 around the ligand are highlighted as red letters. Residues of hLRH-1 that are important for ligand binding are labeled on top of the sequences. c 3D architecture presentation of LRH-1 LBD domain (PDB: 6OQY). LRH-1 contains an additional layer formed by a rigid and relatively long helix H2 (yellow). H12 (green) packs against H3 (green) and H4 (green) to form the AF-2 surface where the co-activator (purple) binds. The agonist (cyan) is depicted as sticks. d Electrostatic potential mapped onto the LBD of hLRH-1 (PDB: 6OQY). Electrostatic map is generated via Pymol and display with gradient color. The agonist (cyan) is depicted as sticks.
A/B and C domains
In the N-terminal region of NR5 members, the A/B structural domain lacks the ligand-independent activation function domain AF-1 [7, 15]. Within the C region of the DBD domain, there is a C-terminal extension (CTE), which establishes additional base-specific contacts within the minor groove of DNA. Adjacent to the distal end of CTE, there also exists a subtype-specific helix known as Ftz-F1, assisting in stabilizing the interaction between LBD and DBD. These two unique structural fragments, CTE and Ftz-F1, form the important structural bases for the unique biochemical activities of LRH-1 and SF-1.
The crystal structure of human LRH-1 solved by Solomon et al. demonstrated that the CTE can recognize DNA response elements that are three base-pairs longer than the standard hexameric sequences [16]. The extension sequence (TCA in human LRH-1) forms five base specific interactions with residues RGGR (162–165) in the CTE segment, facilitating the DBD of LRH-1 to bind the hormone response element (HRE) as a monomer rather than a dimer (for LRH-1, the HRE refers to SFRE, which is the same as SF-1) [17].
Ftz-F1 is a segment of alpha-helix that begins after CTE (Phe168) and is characteristically conserved in the NR5A receptor, forming a helical structure that tightly interacts with the core zinc finger of DBD. Solomon and colleagues designed a mutation experiment to emphasize the importance of the Ftz-F1 helix in the transcriptional regulation of human LRH-1 [16]. Recently, Seacrist et al. established a comprehensive model of full-length LRH-1 using HDX-MS, XL-MS and SAXS experimental data. The model revealed a crucial interaction between the DBD Ftz-F1 helix and the LBD H2, which is facilitated by a salt bridge formed by Arg174 and Asp314. Considering H2 plays a critical role in the ligand-independent activation of LRH-1, the above results suggest a potential link between Ftz-F1 and the ligand-independent activation of LRH-1 [17].
Ligand binding domain (LBD)
The LBD of LRH-1 comprises 12 alpha helices. Three of these helical (H1, H2, and H3) sequences are highly conserved. Additionally, two β strands are formed between helices H5 and H6 (Fig. 1c). For most ligand-dependent receptors, helix 12 (which includes the AF-2 domain) can accommodate various ligands, such as agonist, antagonist, and inverse agonist, resulting in diverse conformations [1]. These conformational changes affect co-the recruitment and dissociation of regulator. Nevertheless, it has been demonstrated that LRH-1 can maintain an active conformation without ligand binding, a phenomenon referred to as constitutive activity [18]. Although the precise mechanisms of action between LRH-1 receptors, its ligands, and co-regulator peptides have not been fully elucidated yet, some co-crystal structures have provided insights into the structural basis of LRH-1’s functional activity.
A typical NR LBD exhibits a common structural motif with a three-layered fold comprising approximately 12 alpha-helices and 2–3 β-strands. However, LRH-1 deviates from this pattern by possessing an additional fourth layer formed by a rigid and relatively long helix H2 (Fig. 1c). A study conducted by Sablin et al. revealed that the seven structural elements defining the ligand binding pocket (LBP) interact with H2. Four of them directly interact with H2 and the remaining three interact indirectly with H2 via H3 [19, 20]. Among those seven elements, H12 packs against H3 and H4 to form the AF-2 surface, which is critical for the determination of the LRH-1 active site (Fig. 1c). Most NRs undergo a conformational change in the ligand-binding pocket upon binding to the ligand, leading to stabilization of the receptor coactivator-binding interface, the AF-2 surface, which results in the formation of a charge clamp that facilitates coactivator recruitment and binding [21]. However, for LRH-1, even in the absence of the ligand, H2 is in close proximity to H12, thus constituting a stable AF-2, which facilitates coactivator binding [22].
LRH-1 is classified as an orphan nuclear receptor due to the lack of identified endogenous ligands [3]. Recent crystallography and mass spectrometry studies have revealed the presence of a large and hydrophobic pocket occupied by certain phospholipids, such as dilauroyl phosphatidylcholine (DLPC) [23, 24]. During the past decade, researchers have discovered a series of synthetic agonists based on a hexahydropentalen scaffold. These agonists share a common binding conformation that the cyclic hydrocarbon backbone fills the large and hydrophobic LBP, and polar groups (-OH, -NH2, etc.) anchor to the polar region either directly or through a hydrogen bonding network of water (Fig. 1d). Additionally, an aromatic substituent forms a stable edge-to-face π-π stacking interaction with His390 [25,26,27]. However, GSK8470, an initial hexahydropentalen agonist of LRH-1, exhibits a unique binding mode due to its ability to form the face-to-face π-π stacking with His390 and the absence of polar groups [25]. This exception suggests the possibility of various binding conformations for GSK8470 analogs, considering the merely 37% occupation of the LRH-1 pocket. Recently, Ortlund’s group identified a series of hybrid phospholipid mimics. Co-crystal structure and molecular dynamics (MD) studies revealed that, beyond deep pocket interactions, a long-chain carboxylic acid in compound interacts with polar residues at the entrance of the pocket, especially Gly421, located in the loop between H6 and H7. This interaction facilitates communication between AF-B to AF-2, leading to signaling similar to that of PLs [28,29,30]. Unfortunately, the co-crystal structure of LRH-1 bound to antagonist has not been reported so far, and the true inactive conformation of LRH-1 and the conformational dynamics of apo-LRH-1 remain unknown. This poses a challenge for the design of LRH-1 antagonists.
Co-regulators play a crucial role in the modulation of the LRH-1 transcriptional activity (Fig. 2) [31]. There are two co-regulator categories consisting of co-activator and co-repressor, comprising approximately 200 different proteins [4, 32]. Currently known coactivators of LRH-1 include SRC, PGC1α, β-catenin and MBF-1, while corepressor comprise SHP, DAX-1, Prox1, NCoR1 and NCoR2. LRH-1 recognizes an α-helix containing a short LXXLL motif (L: Leucine, X: any amino acid) on the co-regulator via an AF-2 surface located in H12 of its LBD [33,34,35]. The leucine residues are situated within the hydrophobic groove of the AF-2 surface, and the ends of the helical peptide promote or inhibit, depending on either co-activator or co-repressor, the charge clamp formed by a lysine on the NR’s H3 and a glutamate on H12. This charge clamp interaction is crucial for modulation of LRH-1’s activity [31, 36, 37].

When LRH-1 recruits transcriptional co-activators, the complex binds to the SFRE of the target gene, activating the expression of downstream target genes. When LRH-1 recruits transcriptional co-repressors, the complex binds to the SFRE of the target gene, inhibiting the expression of downstream target genes.
LRH-1 and diseases
LRH-1 plays multiple roles in development, differentiation, and metabolism. Its expression begins in the early stages of embryonic development and plays a crucial role in embryonic stem cells [38, 39]. Studies on homozygous LRH-1 knockout mice have observed embryonic lethality, further confirming the importance of LRH-1 throughout the development process [40].
In adult mammals, LRH-1 is highly expressed in the liver, pancreas, intestines, steroidogenic organs (adrenal glands), and sex glands (testes and ovaries). It plays a crucial role in regulating cholesterol, bile acid reproduction, as well as steroid hormone homeostasis [7, 41]. In different tissues of the human, LRH-1 regulates diverse transcriptional programs related to metabolism, inflammation, and cell proliferation. These programs include cholesterol efflux, se phosphorylation and transport, bile acid synthesis, lipogenesis, intestinal glucocorticoid synthesis, pancreatic development and differentiation, and androgen synthesis in the sex glands. Additionally, LRH-1 also plays an important role in the energy metabolism of brown adipocytes and high-energy-demanding cardiac muscle cells [42].
Furthermore, LRH-1 is closely associated with the development of various cancers, including colorectal cancer (CRC), pancreatic cancer, prostate cancer, and breast cancer [43,44,45]. The abnormal expression of LRH-1 may lead to malignant transformation and tumor progression. LRH-1 promotes tumor cell proliferation and growth through regulating cancer cell metabolic reprogramming and apoptosis signaling pathways. Consequently, LRH-1 enhances cancer cell survival and promotes cell migration and invasion.
In summary, LRH-1 is essential to many physiological processes, making it crucial for maintaining overall health and normal function. Aberrant expression of LRH-1 has been linked to the development of various diseases, highlighting its potential role in disease pathogenesis (Fig. 3).

LRH-1 regulates the expression of multiple genes associated with cancers, metabolic disorders and inflammation.
LRH-1 and cancers
Studies have shown that microRNAs (miRNAs) participate in the malignant behavior of colorectal cancer cells by directly targeting multiple tumor suppressor genes or oncogenes [46,47,48]. It has been reported that the overexpression of miRNAs such as miR-374B, miR-381, and miR-136, targeting the LRH-1 gene, can inhibit the proliferation and invasion of CRC cells. However, these miRNAs are significantly downregulated in human CRC tissues, with expression levels significantly lower than those in the normal control group [49,50,51,52].
The research conducted by Kramer and colleagues has revealed the mechanism by which LRH-1 promotes CRC growth. Their findings suggest that LRH-1 promotes cell growth by regulating the expression of the cell cycle inhibitor p21 in human CRC cells (HCT116) with wild-type p53. However, in HT29 cells with mutated p53, the regulatory effect of LRH-1 is inhibited, leading to no observed regulation of p21. The results showed that LRH-1 inhibits the recruitment of p53 to the p21 promoter by interacting with co-repressor factor, which allows CRC cells to evade cell cycle arrest mediated by p21 (Fig. 3) [44].
Tumor stem cells play important roles in tumor initiation, progression, and recurrence. Studies have shown that in most CRC cells, overexpression of GATA6 induces upregulation of LRH-1, promoting the expression of stemness characteristics in HCT-116 and HT-29 cells. Additionally, a phenomenon known as the ‘reverse Warburg effect’ has been observed in human CRC stem cells (Fig. 3) [53, 54]. Specifically, overexpression of GATA6 leads to upregulation of LRH-1, significantly enhancing the expression level and activity of HIF-1a. This enhancement stimulates the glycolysis in CRC cells, resulting in the production of a large amount of lactate. Subsequently, lactate is converted to pyruvate by lactate dehydrogenase and is utilized by another subgroup of CRC cells for mitochondrial oxidative phosphorylation. Through this process, metabolic symbiosis and energy transfer are achieved between two subgroups of CRC cells [53].
Furthermore, LRH-1 contributes to CRC development through multiple mechanisms. It regulates glucocorticoid synthesis within CRC cells, impacting the cell cycle and inflammatory pathways. This combined effect promotes the occurrence of intestinal tumors [55, 56]. These findings provided important insights into understanding the occurrence and development of colorectal tumors, thereby contributing to the development of therapeutic strategies targeting LRH-1.
LRH-1 is highly expressed in prostate cancer and promotes de novo synthesis of androgens by activating several key steroidogenic enzymes [45]. This leads to an increase in intracellular androgen levels and androgen receptor signaling intensity, thereby deepening the castration resistance of prostate cancer (Fig. 3). However, LRH-1 antagonists can attenuate the castration resistance of prostate cancer, suggesting LRH-1 can serve as a potential target for the treatment of prostate cancer.
In breast cancer, studies have demonstrated that LRH-1 is widely expressed and plays an important role in the development of the disease. Yasuhiro Miki et al. conducted immunolocalization detection of LRH-1 in 106 human breast cancer tissues [57]. The study results showed that 43% of invasive ductal carcinoma cells expressed LRH-1, and LRH-1 immunoreactivity was also detected in 28% of ductal carcinoma in situ cases. Furthermore, they found a correlation between LRH-1 expression and overall survival in breast cancer patients, especially in the progesterone receptor (PR)-positive breast cancer group (Fig. 3).
In most breast cancers patients, the expression of estrogen receptor-α (ERα) is common. Inhibition of estrogen receptor function is crucial for breast cancer treatment. Thiruchelvam et al. found that LRH-1 regulates the expression of ER by directly binding the ER gene promoter, thereby regulating the growth of breast cancer cells [58]. Additionally, they discovered that LRH-1 is a key regulatory factor in modulating estrogen response in breast cancer, which can regulate estrogen synthesis in breast tumor tissues. In brief, LRH-1 exerts its effects by regulating ER expression and controlling estrogen synthesis in breast tumor tissues (Fig. 3).
Annicotte et al. discovered that the expression of ERα also affects LRH-1 expression [59]. LRH-1 expression is upregulated in ERα-expressing cell lines, while it is downregulated or absent in cell lines that do not express ERα. They also observed a significant enhancement of LRH-1 expression in MCF7 cells after treatment with 17β-estradiol. It is noteworthy that ERα can directly or indirectly regulate the expression of LRH-1, thereby affecting the level of cyclin D1. Cyclin D1 establishes a positive feedback loop by enhancing the transcriptional activity of ERα, further increasing LRH-1 expression, and ultimately leading to increased cell proliferation and tumor progression. ERα plays a regulatory role in LRH-1 expression in breast cancer cells, which mediates the effects of estrogen on breast cancer cell proliferation (Fig. 3). Inhibiting LRH-1 affects the cell proliferation capabilities of different types of breast cancer cells, including ERα-positive and triple-negative cells [60]. Furthermore, LRH-1 modulation in transgenic animal mammary glands can cause changes in mammary morphology by enhancing the expression of transforming growth factor-beta [61]. In summary, the regulation of LRH-1 has multifaceted effects on breast cancer, including modulation of mammary morphology, regulation of cell proliferation, migration, invasion, and chemotherapy resistance.
LRH-1 and metabolic diseases
LRH-1, as an important regulatory factor, holds significant potential for the treatment of diabetes. By targeting LRH-1, the function of pancreatic islet cells can be regulated, and blood glucose control can be restored in different types of type 1 diabetes mouse models [62]. Research has demonstrated that the LRH-1 agonist BL001 exhibits multiple beneficial effects. Firstly, it can prevent the progression of hyperglycemia in type 1 diabetes mouse models and reduce the occurrence of immune-dependent inflammation. Secondly, BL001 can also reduce apoptosis of beta cells in the islets of type 2 diabetes patients, enhancing insulin secretion and the survival rate of beta cells in heterologous transplantation [63]. Additionally, LRH-1 agonist BL001 can activate the LRH-1 and downstream Ptgs2/PGE2/PTGER1 signaling axis, playing an important role in the response of pancreatic islet cells to cytokines, wound healing, and inflammation regulation (Fig. 3) [64].
Baquié et al. discovered that estrogen can increase the expression of LRH-1, thereby protecting pancreatic islet cells [65]. Further research has revealed that estrogen regulates the transcriptional level of LRH-1 through the ERα signaling pathway. Inhibiting LRH-1 can block the protective effect of estrogen on pancreatic islet cell factors. Moreover, glucocorticoids have anti-inflammatory and anti-stress properties, which can alleviate the pro-inflammatory response of pancreatic islet cells. LRH-1 may regulate the glucocorticoids synthesis to attenuate the pro-inflammatory response of the islets, thus protecting them from stress-induced apoptosis (Fig. 3). These research findings emphasize the important role of LRH-1 in the protection of pancreatic islet cells [65].
Recent studies have also found that the expression of LRH-1 and glutaminase 2 (GLS2) are downregulated in diabetic podocytes. However, LRH-1 is activated by administering the LRH-1 agonist DLPC, promoting GLS2-mediated glutamine degradation, thereby improving mitochondrial function and apoptosis in podocytes (Fig. 3). These results support the critical regulatory role of LRH-1 in the energy metabolism of podocytes [66]. Therefore, by modulating the LRH-1 activity it is possible to improve blood glucose control and promote the survival and functional recovery of pancreatic islet cells, providing new avenues for the treatment of diabetes.
LRH-1 is a potential drug target for the treatment of liver diseases. It protects the liver from diet-induced steatosis (fatty liver) and insulin resistance. Studies have shown that mice deficient in LRH-1 exhibit reduced flux of glucokinase (GCK) and glycogen synthase in the liver, suggesting that LRH-1 directly regulates the transcription of GCK. Additionally, the absence of LRH-1 in the liver leads to impaired glucose phosphorylation mediated by GCK, affecting glucose metabolism processes such as glycogen synthesis, glycolysis, and de novo lipogenesis. Therefore, LRH-1 is a critical regulatory factor in hepatic glucose metabolism, controlling glucose uptake and metabolism, and integrating glucose and lipid balance in the postprandial phase (Fig. 3) [67].
Miranda et al. demonstrated that a high-fat diet induces hepatic steatosis, injury, and impaired glucose tolerance in LRH-1-deficienct mice [68]. However, these issues can be reversed by reintroducing wild-type human LRH-1. Further investigation revealed that LRH-1 deficiency inhibits the expression of key genes (Elov15 and Fads2) involved in the biosynthesis of arachidonic acid (AA) phospholipids in the liver, resulting in decreased levels of AA phospholipids (Fig. 3). This highlights the important role of LRH-1 as a phospholipid sensor in maintaining a healthy AA phospholipid pool in the liver.
Moreover, research by Stein et al. revealed that mice with a mutated LRH-1 protein (LRH-1 K289R) exhibited early signs of non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) after being fed a high-fat, high-sugar diet [69]. The LRH-1 K289R mutation led to overexpression of the Osbpl3 gene, promoting the activation of SREBP-1 and stimulating fatty acid synthesis. Silencing the Osbpl3 gene in the liver of these mice successfully reversed the abnormal fatty acid synthesis. This study highlighted the role of LRH-1 protein SUMOylation defect in the development of NAFLD (Fig. 3). In conclusion, LRH-1 plays a crucial role in the liver by regulating glucose metabolism, maintaining a healthy AA phospholipid pool, and preventing the development of NAFLD. These research findings strongly suggest LRH-1 as a promising drug target for the treatment of liver diseases.
LRH-1 and inflammations
LRH-1 plays a critical role in inflammatory bowel disease (IBD). IBD is characterized by epithelial dysfunction and crypt damage, and LRH-1 is crucial for maintaining intestinal epithelial health and protecting against inflammatory damage. Studies have found that LRH-1 knockout leads to adverse effects such as decreased Notch signaling, increased crypt cell death, distorted epithelial cell composition, and weakened epithelial barrier function. Conversely, overexpression of LRH-1 can restore epithelial integrity and alleviate inflammatory damage in the intestinal organs, including those of IBD patients (Fig. 3). Futhermore, LRH-1 can reduce the severity of T cell-mediated colitis in mice. These findings highlight the important role of LRH-1 in promoting normal intestinal epithelial cell dynamics and protecting the intestine from inflammation [70].
Research has demonstrated that haploinsufficiency of LRH-1 increases the risk of intestinal inflammation in mice and leads to enhanced inflammatory responses. When LRH-1 heterozygous mice are exposed to 2,4,6-trinitrobenzene sulfonic acid, it impairs the expression of Cyp11a1 and Cyp11b1, which control local corticosterone synthesis in the intestine, resulting in reduced local corticosterone production. Further studies have revealed that mice lacking epithelial-specific LRH-1 display reduced levels of local glucocorticoids. Moreover, colonic biopsies from patients with Crohn’s disease and ulcerative colitis also show reduced expression of LRH-1 and genes involved in glucocorticoid production (Fig. 3). Therefore, LRH-1 regulates intestinal immunity by promoting local glucocorticoid production, playing a significant role in immune stress, and emphasizing its importance in controlling intestinal inflammation and the pathogenesis of IBD [71].
Ahmed et al. discovered a strong association between LRH-1 and the expression of steroidogenic enzymes. In colitis-induced mouse colon, loss of LRH-1 significantly reduces the expression of steroidogenic enzymes. Similarly, colonic biopsies from patients with IBD show reduced expression of the steroidogenic enzyme HSD11B1, which may be associated with defective local activation of glucocorticoids (Fig. 3) [72]. Furthermore, Landskron et al. demonstrated that glucocorticoid receptors can induce LRH-1 to participate in intestinal steroid hormone production [73].
In conclusion, LRH-1 plays a critical role in IBD, maintaining intestinal epithelial health, and protecting against inflammatory damage. Loss of LRH-1 leads to epithelial dysfunction, crypt damage, and increased immune stress. LRH-1 is closely associated with the expression of steroidogenic enzymes and the production of glucocorticoid.
LRH-1 modulators
The nuclear receptor LRH-1, a transcription factor, plays an important role in embryonic development, steroid and cholesterol metabolism, inflammation, and multiple types of cancer [45, 74,75,76,77,78]. LRH-1 has constitutive activity and is considered an orphan nuclear receptor. Researchers have been actively exploring the development of small molecule modulators to target LRH-1. Crystallographic studies have demonstrated that various phospholipids can bind to the ligand-binding pocket of human LRH-1, indicating the potential for small molecule modulation of LRH-1 activity. However, the designs of ligands for LRH-1 remain highly comlex due to the strong hydrophobicity and large space of the ligand-binding pocket of LRH-1.
Despite these challenges, scientists have made significant progress in the discovery of LRH-1 modulators. The identification and development of small molecules that can specifically bind to and modulate LRH-1 activity have the potential to provide valuable insights into LRH-1’s biological functions and therapeutic applications.
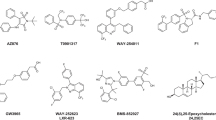
In 2006, Whitby and his colleagues discovered the first LRH-1 small molecule ligand, GSK8470 [79]. It has a hydrophobic chemical motif consisting of a cis-fused bicyclo[3.3.0]-octene core that provides rigidity, low molecular weight, and the potential for three-position functionalization. This structural design makes it suitable for the development of novel nuclear receptor ligands.
Using FRET technology, the researchers observed that GSK8470 can activate the transcriptional activity of LRH-1 (IC50 = 430 nM, Table 1). Furthermore, GSK8470 increases the expression of the LRH-1 target gene small heterodimer partner (SHP) in liver cells. Co-crystal structure revealed that the phenyl group of GSK8470 forms face-to-face π-π interaction with His390 (Fig. 4a). However, the 1-phenylamine moiety in GSK8470 exhibits acid instability, with a half-life of approximately 12 h in the presence of 1 M acetic acid. Therefore, further modifications are needed to improve its acid stability and oral bioavailability [79]. The discovery of GSK8470 as an LRH-1 agonist represents a significant milestone in the development of LRH-1 modulators. However, further research and modification are necessary to overcome limitations for potential therapeutic applications.

a GSK8470 (PDB: 3PLZ, purple) forms a face-to-face π-π stack with His390. b RJW100 (PDB: 5L11, green) forms water-mediated hydrogen bonds with Asp389, His390, Arg393 and Thr352, and an edge-to-face π-π stack with His390. c 6N (PDB: 6OQY, light brown) forms direct hydrogen bonds with Met345 and Thr352, water-mediated hydrogen bonds with His390 and Arg393, and an edge-to-face π-π stack with His390. d 6N-10CA (PDB: 7TT8, pink) forms direct hydrogen bonds with Met345 and Thr352, water-mediated hydrogen bonds with His390 and Arg393, and an edge-to-face π-π stack with His390 deep within the pocket. Meanwhile, its carboxyl tail also forms direct or water-mediated hydrogen bonds with Gly421, Leu424, Tyr516 and Lys520.
In 2011, Whitby and colleagues further optimized the structure of GSK8470 and obtained a series of more potent compounds RJW100-RJW103, all of which feature a hexahydropentalene core structure [80].
Due to the lack of polar interaction between the acid-sensitive phenylamine nitrogen in GSK8470 and proteins, the researchers replaced the phenylamine with oxygen and carbon atoms. Moreover, incorporating polar moieties capable of interacting with residues His390, Arg393, and Gln432 within a 5 Å range into GSK8470 potentially enhanced both the binding affinity and ligand hydrophilicity. Two new classes of compounds identified based on these two strategies were determined through FRET experiments. Both classes of compounds exhibited good activity, with RJW100 acting as a dual LRH-1 and SF-1 agonist (pEC50 = 6.6 for LRH-1 and pEC50 = 7.5 for SF-1). The co-crystal structure of RJW100 with LRH-1 showed that the hydroxyl group of RJW100 can interact with Asp389, His390, Arg393 and Thr352 through water molecules (Fig. 4b). These interactions enhance its binding activity with the LRH-1 LBP and lead to a distinct binding conformation compared to GSK8470, which changed the original face-to-face π-π stack into an edge-to-face π-π stack. RJW101 demonstrated selective LRH-1 agonist activity, while compounds RJW102 and RJW103 showed SF-1 agonist activities (Table 1) [80].
The development of the RJW series of compounds represents an advancement in the optimization of LRH-1 modulators. By incorporating structural modifications and targeting specific residues, these compounds showed improved potency and selectivity as LRH-1 and SF-1 agonists.
LRH-1 has been shown to interact with various phospholipids [81, 82]. Among them, DLPC can activate LRH-1 both in vitro and in vivo. DLPC was found to have anti-diabetic effects by Lee and colleagues in 2011 [83]. Flynn et al. identified that the phosphate head of DLPC can interact with the polar amino acid residues near the entrance of the LBP, causing conformational changes in LRH-1 LBD and facilitating the transmission of allosteric signals from the LBP to the activation functional surface [84].
On the other hand, the binding of RJW100 to LRH-1 is primarily mediated by the interaction between the hydroxyl group and Thr352. The researchers further overlapped the co-crystal structures of RJW100 and DLPC bound to LRH-1 LBP, and observed that the hexyl tail of RJW100 overlaps with the lauryl tail of DLPC. Therefore, Flynn and colleagues designed a series of hybrid phospholipid analogs by merging key groups from both RJW100 and DLPC. These analogs have three distinct regions: the 6HP core of RJW100 interacts with Thr352, the alkyl chain maintains the hydrophobicity, and the terminal polar group forms water-mediated hydrogen bond networks with the residues near the pocket entrance [84].
Considering the potential limitations of phosphates, such as their poor cell permeability and low stability, the researchers replaced the phosphate ester of the hybrid phospholipid analogs with a carboxylic acid. This modification resulted in a more potent LRH-1 agonist 6HP-CA (IC50 = 400 nM, Table 1) [84].
Mays and colleagues conducted the crystal structure analysis of LRH-1-RJW100 and discovered that the outer hydroxyl group of the ligand can forms hydrogen bonds with polar residues (Thr352, His390, Arg393) through a water network deep in the LBP. To further investigate the importance of this polar interaction in activating LRH-1 by RJW100, the researchers used an RJW100 analog lacking the hydroxyl group and introduced a Thr352Val mutation in LRH-1. Through these experiments, they demonstrated the essential role of the polar interaction in the activation of LRH-1 by RJW100. Building upon this, they replaced the bridging water molecule with larger polar groups, which can directly interact with Thr352 or other nearby polar amino acid residues. Meanwhile, they retained the edge-to-face π-π stack with His390. This modification aimed to achieve a more predictably and stable ligand conformation in the desired region of the pocket. Through these efforts, they successfully obtained a potent LRH-1 agonist 6N (IC50 = 15 nM) and a co-crystal structure of 6N with LRH-1 (Fig. 4c). Notably, 6N exhibited a 100-fold improvement in activity compared to RJW100 (Table 1) [85].
In addition to its enhanced potency, 6N demonstrated promising therapeutic potential in an organic inflammatory bowel disease (IBD) model. It upregulated the expression of steroidogenic genes controlled by LRH-1 and induced changes in the expression of anti-inflammatory genes [85].
Cato and colleagues investigated the role of binding sites within the LBP of LRH-1 on ligand affinity, potency, and protein activity. They found that both the deep and entrance binding sites of the LBP of LRH-1 contribute to ligand affinity and potency, with an additive effect. Therefore, based on RJW100, the researchers aimed to optimize the ligand binding to the deep and entrance regions of the pocket. This strategy leads to the development of compounds 6N and 10CA. Combining the advantages of these two compounds, they further obtained compound 6N-10CA (EC50 = 43 nM) that exhibited high affinity, potency, and drug efficacy (Table 1). 6N-10CA forms direct hydrogen bonds with Met345 and Thr352, water-mediated hydrogen bonds with His390 and Arg393, and edge-to-face π-π stack deep within the pocket. Meanwhile, its carboxyl tail also forms direct or water-mediated hydrogen bonds with Gly421, Leu424, Tyr516 and Lys520. These interactions significantly increase the affinity of 6N-10CA (Fig. 4d) [29].
To gain a comprehensive understanding of the LRH-1 agonist 6N-10CA, subsequent multidimensional studies were conducted, including gene expression, co-regulator binding and signal transduction, co-crystal structure, and dynamics of ligand binding to the LRH-1 pocket. The results showed that binding to the deep pocket of LRH-1 confers high affinity, while binding to the entrance of the pocket dominates the allosteric effect and transcriptional activity of lipid-like compounds [29].
The discovery and development of 6N-10CA, a highly potent LRH-1 agonist, provide valuable insights into the structural and functional aspects of LRH-1 modulation. Further research is needed to explore the therapeutic potential of 6N-10CA and its applications in the treatment of inflammatory diseases and other disorders.
Studies have shown that LRH-1 plays a significant role in tumor formation both in vitro and in vivo. Therefore, the search for potent and selective LRH-1 antagonists may provide new drug candidate for cancer treatment. ML179 and ML180 are the first LRH-1 inverse agonists and have made important contributions in this field [22].
While the IC50 of ML180 on LRH-1 is only 3.7 μM, it exhibits the maximum inhibitory effect on LRH-1, reaching 64% at a concentration of 10 μM. ML179 demonstrates potent inhibitory activity against LRH-1 with an IC50 of 0.32 μM. However, compared to ML180, its maximum inhibitory effect on LRH-1 is only 40%. Research suggests that ML180 and ML179 have shown promising results in suppressing cancer cell growth, particularly exhibiting higher activity in multiple breast cancer cell lines [22, 86].
The Benod et al. conducted molecular docking screening of 5.2 million commercially available compounds to identify potential LRH-1 antagonists [87]. The top-ranked molecules were further evaluated through in vitro binding experiments and transcriptional activity assays. Biological evaluations discovered that compounds Cpd3 and Cpd3d2 effectively inhibit the transcriptional activity of LRH-1 and reduce the expression of target genes, including the co-transcriptional repressor factor SHP, cyclin E1 and G0S2, which are involved in cell growth and proliferation.
Cpd3 and Cpd3d2 act as selective LRH-1 antagonists, binding to the receptor and inhibiting the receptor-mediated transcriptional activity with IC50 values of 5 ± 1 µM and 6 ± 1 µM, respectively. Moreover, Cpd3 and Cpd3d2 effectively suppress the growth of pancreatic cancer cells, colon cancer cells, and breast cancer cells (Table 1). Importantly, both compounds did not exhibit significant toxicity towards normal cells even at high concentrations. This characteristic makes them valuable molecular tools for researchers to elucidate the action mechanisms of LRH-1 in different cellular environments [87].
Their abilities to inhibit LRH-1 transcriptional activity and suppress the growth of cancer cells highlight their potential as therapeutic agents for various types of cancer.
Summary and perspectives
In summary, LRH-1, an orphan nuclear receptor, has emerged as a promising drug target with significant implications for treating various human diseases. Recent studies have revealed the crucial role of LRH-1 in regulating transcriptional processes related to metabolism, inflammation, and cell proliferation in different tissues of the human body. LRH-1 is involved in key physiological processes including cholesterol transport, glucose metabolism, bile acid synthesis, lipogenesis, gut hormone synthesis, pancreatic development and differentiation, and sex hormone production.
LRH-1’s regulatory role in metabolism makes it a promising target for the treatment of metabolic disorders like non-alcoholic fatty liver disease and diabetes. The overexpression of LRH-1 has been shown to alleviate disease-associated inflammation and cell death, suggesting the therapeutic potential of LRH-1 in IBD. Furthermore, LRH-1 is considered as an important target for the treatment of estrogen receptor-positive breast cancer, prostate cancer, colon cancer, pancreatic cancer, and other cancers. LRH-1 ligands have the potential to be developed as drug candidates for the treatment of human diseases, such as IBD, fatty liver disease, diabetes and cancers.
In conclusion, LRH-1 has emerged as a potential drug target with significant implications for treating various human diseases. Future research and development efforts should focus on elucidating the mechanisms of LRH-1 and strive to develop more potent and selective LRH-1 small molecule modulators that can be translated into effective clinical applications, ultimately benefiting patients.
References
Zhang Y, Luo XY, Wu DH, Xu Y. ROR nuclear receptors: structures, related diseases, and drug discovery. Acta Pharmacol Sin. 2015;36:71–87.
Wärnmark A, Treuter E, Wright AP, Gustafsson JA. Activation functions 1 and 2 of nuclear receptors: molecular strategies for transcriptional activation. Mol Endocrinol. 2003;17:1901–9.
Sar P. Nuclear receptor: structure and function. Prog Mol Biol Transl Sci. 2023;196:209–27.
Weikum ER, Liu X, Ortlund EA. The nuclear receptor superfamily: a structural perspective. Protein Sci. 2018;27:1876–92.
Giguère V. Orphan nuclear receptors: from gene to function. Endocr Rev. 1999;20:689–725.
Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–96.
Fayard E, Auwerx J, Schoonjans K. LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004;14:250–60.
Zerlotin R, Arconzo M, Piccinin E, Moschetta A. Another one bites the gut: nuclear receptor LRH-1 in intestinal regeneration and cancer. Cancers. 2021;13:896.
Sun Y, Demagny H, Schoonjans K. Emerging functions of the nuclear receptor LRH-1 in liver physiology and pathology. Biochim Biophys Acta Mol Basis Dis. 2021;1867:166145.
Michalek S, Brunner T. Nuclear-mitochondrial crosstalk: on the role of the nuclear receptor liver receptor homolog-1 (NR5A2) in the regulation of mitochondrial metabolism, cell survival, and cancer. IUBMB Life. 2021;73:592–610.
Yazawa T, Imamichi Y, Miyamoto K, Khan MR, Uwada J, Umezawa A, et al. Regulation of steroidogenesis, development, and cell differentiation by steroidogenic factor-1 and liver receptor homolog-1. Zool Sci. 2015;32:323–30.
Mouzat K, Baron S, Marceau G, Caira F, Sapin V, Volle DH, et al. Emerging roles for LXRs and LRH-1 in female reproduction. Mol Cell Endocrinol. 2013;368:47–58.
Sandhu N, Rana S, Meena K. Nuclear receptor subfamily 5 group A member 2 (NR5A2): role in health and diseases. Mol Biol Rep. 2021;48:8155–70.
Meinsohn M-C, Smith OE, Bertolin K, Murphy BD. The orphan nuclear receptors steroidogenic factor-1 and liver receptor homolog-1: structure, regulation, and essential roles in mammalian reproduction. Physiol Rev. 2019;99:1249–79.
Li LA, Chiang EFL, Chen JC, Hsu NC, Chen YJ, Chung BC. Function of steroidogenic factor 1 domains in nuclear localization, transactivation, and interaction with transcription factor TFIIB and c-Jun. Mol Endocrinol. 1999;13:1588–98.
Solomon IH, Hager JM, Safi R, McDonnell DP, Redinbo MR, Ortlund EA. Crystal structure of the human LRH-1 DBD-DNA complex reveals Ftz-F1 domain positioning is required for receptor activity. J Mol Biol. 2005;354:1091–102.
Seacrist CD, Kuenze G, Hoffmann RM, Moeller BE, Burke JE, Meiler J, et al. Integrated structural modeling of full-length LRH-1 reveals inter-domain interactions contribute to receptor structure and function. Structure. 2020;28:830–46.
Lu Y, Anderson WR, Zhang H, Feng S, Pick L. Functional conservation of drosophila FTZ-F1 and its mammalian homologs suggests ligand-independent regulation of NR5A family transcriptional activity. Dev Genes Evol. 2013;223:199–205.
Steinmetz AC, Renaud JP, Moras D. Binding of ligands and activation of transcription by nuclear receptors. Annu Rev Biophys Biomol Struct. 2001;30:329–59.
Sablin EP, Krylova IN, Fletterick RJ, Ingraham HA. Structural basis for ligand-independent activation of the orphan nuclear receptor LRH-1. Mol Cell. 2003;11:1575–85.
Nagy L, Schwabe JW. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci. 2004;29:317–24.
Busby S, Nuhant P, Cameron M, Mercer BA, Hodder P, Roush WR, et al. Discovery of inverse agonists for the liver receptor homologue-1 (LRH1; NR5A2). Probe reports from the NIH molecular libraries program. Bethesda (MD): National Center for Biotechnology Information (US); 2010.
Forman BM. Are those phospholipids in your pocket? Cell Metab. 2005;1:153–55.
Xu D, Jiang X, Wang Y, Song S. Liver receptor homolog-1 regulates apoptosis of bovineovarian granulosa cells by progestogen receptor signaling pathway. Animals. 2022;12:1213.
Mays SG, Okafor CD, Whitby RJ, Goswami D, Stec J, Flynn AR, et al. Crystal structures of the nuclear receptor, liver receptor homolog 1, bound to synthetic agonists. J Biol Chem. 2016;291:25281–91.
Wu X, Zhang Y, Xu Y. Discovery of the first low nanomolar liver receptor homolog-1 (LRH-1) agonist. J Med Chem. 2019;62:11019–21.
Cornelison JL, Cato ML, Johnson AM, D’Agostino EH, Melchers D, Patel AB, et al. Development of a new class of liver receptor homolog-1 (LRH-1) agonists by photoredox conjugate addition. Bioorg Med Chem Lett. 2020;30:127293.
Cato ML, D’Agostino EH, Spurlin RM, Flynn AR, Cornelison JL, Johnson AM, et al. Comparison of activity, structure, and dynamics of SF-1 and LRH-1 complexed with small molecule modulators. J Biol Chem. 2023;299:104921.
Cato ML, Cornelison JL, Spurlin RM, Courouble VV, Patel AB, Flynn AR, et al. Differential modulation of nuclear receptor LRH-1 through targeting buried and surface regions of the binding pocket. J Med Chem. 2022;65:6888–902.
Mays SG, Hercules D, Ortlund EA, Okafor CD. The nuclear receptor LRH-1 discriminates between ligands using distinct allosteric signaling circuits. Protein Sci. 2023;32:e4754.
Lazarus KA, Wijayakumara D, Chand AL, Simpson ER, Clyne CD. Therapeutic potential of liver receptor homolog-1 modulators. J Steroid Biochem Mol Biol. 2012;130:138–46.
Lonard DM, O’malley BW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell. 2007;27:691–700.
Zhao H, Li Z, Cooney AJ, Lan ZJ. Orphan nuclear receptor function in the ovary. Front Biosci. 2007;12:3398–405.
Suzuki T, Kasahara M, Yoshioka H, Morohashi K, Umesono K. LXXLL-related motifs in Dax-1 have target specificity for the orphan nuclear receptors Ad4BP/SF-1 and LRH-1. Mol Cell Biol. 2003;23:238–49.
Yazawa T, Inaoka Y, Okada R, Mizutani T, Yamazaki Y, Usami Y, et al. PPAR-gamma coactivator-1alpha regulates progesterone production in ovarian granulosa cells with SF-1 and LRH-1. Mol Endocrinol. 2010;24:485–96.
Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, et al. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–56.
Stein S, Schoonjans K. Molecular basis for the regulation of the nuclear receptor LRH-1. Curr Opin Cell Biol. 2015;33:26–34.
Gu P, Goodwin B, Chung AC, Xu X, Wheeler DA, Price RR, et al. Orphan nuclear receptor LRH-1 is required to maintain oct4 expression at the epiblast stage of embryonic development. Mol Cell Biol. 2005;25:3492–505.
Heng JC, Feng B, Han J, Jiang J, Kraus P, Ng JH, et al. The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell. 2010;6:167–74.
Paré JF, Malenfant D, Courtemanche C, Jacob-Wagner M, Roy S, Allard D, et al. The fetoprotein transcription factor (FTF) gene is essential to embryogenesis and cholesterol homeostasis and is regulated by a DR4 element. J Biol Chem. 2004;279:21206–16.
Meinsohn MC, Morin F, Bertolin K, Duggavathi R, Schoonjans K, Murphy BD. The orphan nuclear receptor liver homolog receptor-1 (Nr5a2) regulates ovarian granulosa cell proliferation. J Endocr Soc. 2018;2:24–41.
Higashiyama H, Kinoshita M, Asano S. Expression profiling of liver receptor homologue 1 (LRH-1) in mouse tissues using tissue microarray. J Mol Histol. 2007;38:45–52.
Nadolny C, Dong X. Liver receptor homolog-1 (LRH-1): a potential therapeutic target for cancer. Cancer Biol Ther. 2015;16:997–1004.
Kramer HB, Lai CF, Patel H, Periyasamy M, Lin ML, Feller SM, et al. LRH-1 drives colon cancer cell growth by repressing the expression of the CDKN1A gene in a p53-dependent manner. Nucleic Acids Res. 2016;44:582–94.
Xiao L, Wang Y, Xu K, Hu H, Xu Z, Wu D, et al. Nuclear receptor LRH-1 functions to promote castration-resistant growth of prostate cancer via its promotion of intratumoral androgen biosynthesis. Cancer Res. 2018;78:2205–18.
Faber C, Kirchner T, Hlubek F. The impact of microRNAs on colorectal cancer. Virchows Arch. 2009;454:359–67.
Slaby O, Svoboda M, Michalek J, Vyzula R. MicroRNAs in colorectal cancer: translation of molecular biology into clinical application. Mol Cancer. 2009;8:102.
Schetter AJ, Okayama H, Harris CC. The role of microRNAs in colorectal cancer. Cancer J. 2012;18:244–52.
Qu R, Hao S, Jin X, Shi G, Yu Q, Tong X, et al. MicroRNA-374b reduces the proliferation and invasion of colon cancer cells by regulation of LRH-1/Wnt signaling. Gene. 2018;642:354–61.
Liang Y, Zhao Q, Fan L, Zhang Z, Tan B, Liu Y, et al. Down-regulation of MicroRNA-381 promotes cell proliferation and invasion in colon cancer through up-regulation of LRH-1. Biomed Pharmacother. 2015;75:137–41.
Yuan Q, Cao G, Li J, Zhang Y, Yang W. MicroRNA-136 inhibits colon cancer cell proliferation and invasion through targeting liver receptor homolog-1/Wnt signaling. Gene. 2017;628:48–55.
Yan L, Qiu J, Yao J. Downregulation of microRNA-30d promotes cell proliferation and invasion by targeting LRH-1 in colorectal carcinoma. Int J Mol Med. 2017;39:1371–80.
Lai HT, Chiang CT, Tseng WK, Chao TC, Su Y. GATA6 enhances the stemness of human colon cancer cells by creating a metabolic symbiosis through upregulating LRH-1 expression. Mol Oncol. 2020;14:1327–47.
Wilde L, Roche M, Domingo-Vidal M, Tanson K, Philp N, Curry J, et al. Metabolic coupling and the reverse warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44:198–203.
Sidler D, Renzulli P, Schnoz C, Berger B, Schneider-Jakob S, Flück C, et al. Colon cancer cells produce immunoregulatory glucocorticoids. Oncoimmunology. 2012;1:529–30.
Schoonjans K, Dubuquoy L, Mebis J, Fayard E, Wendling O, Haby C, et al. Liver receptor homolog 1 contributes to intestinal tumor formation through effects on cell cycle and inflammation. Proc Natl Acad Sci USA. 2005;102:2058–62.
Miki Y, Clyne CD, Suzuki T, Moriya T, Shibuya R, Nakamura Y, et al. Immunolocalization of liver receptor homologue-1 (LRH-1) in human breast carcinoma: possible regulator of insitu steroidogenesis. Cancer Lett. 2006;244:24–33.
Thiruchelvam PT, Lai CF, Hua H, Thomas RS, Hurtado A, Hudson W, et al. The liver receptor homolog-1 regulates estrogen receptor expression in breast cancer cells. Breast Cancer Res Treat. 2011;127:385–96.
Annicotte JS, Chavey C, Servant N, Teyssier J, Bardin A, Licznar A, et al. The nuclear receptor liver receptor homolog-1 is an estrogen receptor target gene. Oncogene. 2005;24:8167–75.
Bianco S, Jangal M, Garneau D, Gévry N. LRH-1 controls proliferation in breast tumor cells by regulating CDKN1A gene expression. Oncogene. 2015;34:4509–18.
Lazarus KA, Brown KA, Young MJ, Zhao Z, Coulson RS, Chand AL, et al. Conditional overexpression of liver receptor homolog-1 in female mouse mammary epithelium results in altered mammary morphogenesis via the induction of TGF-β. Endocrinology. 2014;155:1606–17.
Cobo-Vuilleumier N, Lorenzo PI, Gauthier BR. Targeting LRH-1/NR5A2 to treat type 1 diabetes mellitus. Cell Stress. 2018;2:141–43.
Cobo-Vuilleumier N, Lorenzo PI, Rodríguez NG, Herrera Gómez IG, Fuente-Martin E, López-Noriega L, et al. LRH-1 agonism favours an immune-islet dialogue which protects against diabetes mellitus. Nat Commun. 2018;9:1488.
Martin Vázquez E, Cobo-Vuilleumier N, Araujo Legido R, Marín-Cañas S, Nola E, Dorronsoro A, et al. NR5A2/LRH-1 regulates the PTGS2-PGE(2)-PTGER1 pathway contributing to pancreatic islet survival and function. iScience. 2022;25:104345.
Baquié M, St-Onge L, Kerr-Conte J, Cobo-Vuilleumier N, Lorenzo PI, Jimenez Moreno CM, et al. The liver receptor homolog-1 (LRH-1) is expressed in human islets and protects {beta}-cells against stress-induced apoptosis. Hum Mol Genet. 2011;20:2823–33.
Hu J, Zhang Z, Hu H, Yang K, Zhu Z, Yang Q, et al. LRH-1 activation alleviates diabetes-induced podocyte injury by promoting GLS2-mediated glutaminolysis. Cell Prolif. 2023;56:e13479.
Oosterveer MH, Mataki C, Yamamoto H, Harach T, Moullan N, van Dijk TH, et al. LRH-1-dependent glucose sensing determines intermediary metabolism in liver. J Clin Invest. 2012;122:2817–26.
Miranda DA, Krause WC, Cazenave-Gassiot A, Suzawa M, Escusa H, Foo JC, et al. LRH-1 regulates hepatic lipid homeostasis and maintains arachidonoyl phospholipid pools critical for phospholipid diversity. JCI Insight. 2018;3:e96151.
Stein S, Lemos V, Xu P, Demagny H, Wang X, Ryu D, et al. Impaired SUMOylation of nuclear receptor LRH-1 promotes nonalcoholic fatty liver disease. J Clin Invest. 2017;127:583–92.
Bayrer JR, Wang H, Nattiv R, Suzawa M, Escusa HS, Fletterick RJ, et al. LRH-1 mitigates intestinal inflammatory disease by maintaining epithelial homeostasis and cell survival. Nat Commun. 2018;9:4055.
Coste A, Dubuquoy L, Barnouin R, Annicotte JS, Magnier B, Notti M, et al. LRH-1-mediated glucocorticoid synthesis in enterocytes protects against inflammatory bowel disease. Proc Natl Acad Sci USA. 2007;104:13098–103.
Ahmed A, Schwaderer J, Hantusch A, Kolho KL, Brunner T. Intestinal glucocorticoid synthesis enzymes in pediatric inflammatory bowel disease patients. Genes Immun. 2019;20:566–76.
Landskron G, Dubois-Camacho K, Orellana-Serradell O, De la Fuente M, Parada-Venegas D, Bitrán M, et al. Regulation of the intestinal extra-adrenal steroidogenic pathway component LRH-1 by glucocorticoids in ulcerative colitis. Cells. 2022;11:1905.
Benod C, Vinogradova MV, Jouravel N, Kim GE, Fletterick RJ, Sablin EP. Nuclear receptor liver receptor homologue 1 (LRH-1) regulates pancreatic cancer cell growth and proliferation. Proc Natl Acad Sci USA. 2011;108:16927–31.
Sidler D, Renzulli P, Schnoz C, Berger B, Schneider-Jakob S, Flück C, et al. Colon cancer cells produce immunoregulatory glucocorticoids. Oncogene. 2011;30:2411–19.
Zhou J, Suzuki T, Kovacic A, Saito R, Miki Y, Ishida T, et al. Interactions between prostaglandin E(2), liver receptor homologue-1, and aromatase in breast cancer. Cancer Res. 2005;65:657–63.
Wang Z, Wu D, Ng CF, Teoh JY, Yu S, Wang Y, et al. Nuclear receptor profiling in prostatospheroids and castration-resistant prostate cancer. Endocr Relat Cancer. 2018;25:35–50.
Chand AL, Pathirage N, Lazarus K, Chu S, Drummond AE, Fuller PJ, et al. Liver receptor homologue-1 expression in ovarian epithelial and granulosa cell tumours. Steroids. 2013;78:700–06.
Whitby RJ, Dixon S, Maloney PR, Delerive P, Goodwin BJ, Parks DJ, et al. Identification of small molecule agonists of the orphan nuclear receptors liver receptor homolog-1 and steroidogenic factor-1. J Med Chem. 2006;49:6652–55.
Whitby RJ, Stec J, Blind RD, Dixon S, Leesnitzer LM, Orband-Miller LA, et al. Small molecule agonists of the orphan nuclear receptors steroidogenic factor-1 (SF-1, NR5A1) and liver receptor homologue-1 (LRH-1, NR5A2). J Med Chem. 2011;54:2266–81.
Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, et al. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. 2005;120:343–55.
Ortlund EA, Lee Y, Solomon IH, Hager JM, Safi R, Choi Y, et al. Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nat Struct Mol Biol. 2005;12:357–63.
Lee JM, Lee YK, Mamrosh JL, Busby SA, Griffin PR, Pathak MC, et al. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature. 2011;474:506–10.
Flynn AR, Mays SG, Ortlund EA, Jui NT. Development of hybrid phospholipid mimics as effective agonists for liver receptor homologue-1. ACS Med Chem Lett. 2018;9:1051–56.
Mays SG, Flynn AR, Cornelison JL, Okafor CD, Wang H, Wang G, et al. Development of the first low nanomolar liver receptor homolog-1 agonist through structure-guided design. J Med Chem. 2019;62:11022–34.
Corzo CA, Mari Y, Chang MR, Khan T, Kuruvilla D, Nuhant P, et al. Antiproliferation activity of a small molecule repressor of liver receptor homolog 1. Mol Pharmacol. 2015;87:296–304.
Benod C, Carlsson J, Uthayaruban R, Hwang P, Irwin JJ, Doak AK, et al. Structure-based discovery of antagonists of nuclear receptor LRH-1. J Biol Chem. 2013;288:19830–44.
Acknowledgements
We gratefully acknowledge financial support from the National Key Research and Development Plan (grant 2022YFE0210600), the Joint Fund of Shandong Natural Science Foundation (grant ZR2022LSW026), the Science and Technology Program of Guangzhou, China (grant 202201010138).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, T., Lu, Zf., Yu, Hn. et al. Liver receptor homolog-1: structures, related diseases, and drug discovery. Acta Pharmacol Sin (2024). https://doi.org/10.1038/s41401-024-01276-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41401-024-01276-x
