
Abstract
Ten new proansamycin B congeners (1–10) together with one known (11) were isolated and characterized on the basis of 1D and 2D NMR spectroscopic and HRESIMS data from the Amycolatopsis mediterranei S699 ΔPM::rifR+rif-orf19 mutant. Compounds 8 and 9 featured with six-membered ring and five-membered ring hemiketal, respectively. Compounds 1, 2, and 9 displayed antibacterial activity against MRSA (methicillin-resistant Staphylococcus aureus), with the MIC (minimal inhibitory concentration) values of 64, 8, and 128 µg/mL, respectively. Compound 1 showed significant cytotoxicity against MDA-MB-231, HepG2 and Panc-1 cell lines with IC50 (half maximal inhibitory concentration) values of 2.3 ± 0.2, 2.5 ± 0.3 and 3.8 ± 0.5 μM, respectively.
Similar content being viewed by others
Introduction
Ansamycins are a class of macrolactams with significant bioactivities, including anti-tuberculosis rifamycins [1], antitumor maytansinoids [2] and geldanamycins [3]. Ansamycins are synthesized by type I polyketide synthase (PKS) that used 3-amino-5-hydroxybenzoic acid (AHBA) as the initial unit for modular loading and embellished through assorted complex post-PKS modifications [4]. Rifamycins were first discovered in 1957, which exhibited bactericidal activities by inhibiting DNA transcription [5]. However, resistance was common in the process of drug overuse and mainly because of target modification and antibiotic inactivation by pathogens [6,7,8]. Therefore, discovery of new anti-tuberculosis rifamycin derivatives is of particular importance. Five type I polyketide synthases (PKSs: RifA–E) charged for the assemble of undecaketide carbon skeleton, which was then cyclized by the amide synthase RifF to release the first presumable macrocyclic precursor (prorifamycins) [9]. The subsequent post-PKS modifications, which included the dehydrogenation of C-8 hydroxy group and the hydroxylation of C-34 methyl group, resulted in the production of rifamycin W [10, 11]. Then Rif-Orf5 dedicates the transformation from the Δ12,29 olefinic bond of rifamycin W into the ketal. Next, the acetylation of the hydroxy group at C-25 regulated by Rif-Orf20 and the O-methylation at C-27 regulated by Rif-Orf14 produced rifamycin SV (R-SV) [12, 13]. The biosynthesis process from R-SV to rifamycin B (R-B) had been thoroughly studied [14]. In this study, Amycolatopsis mediterranei S699 ΔPM::rifR+rif-orf19 mutant was constructed by deleting post-PKS modification genes to afford ten new congeners of proansamycin B [15] (Fig. 1). These metabolites showed antibacterial and cytotoxic properties. These findings provide direction for the future development of potential anti-MRSA and antitumor drugs by structure modifications of rifamycins based on biosynthesis.

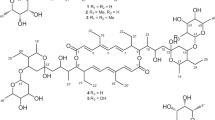
Structures of proansamycin B and congeners 1–11
Results and discussion
To explore the modification of rifamycin structure by extra-cluster genes, two post-PKS modification gene fragments within the rif gene cluster, namely rif-orf3 to rif-orf16 and rif-orf33/rifT/rif-orf0, were deleted to generate the ΔPM mutant. The production of rifamycins was totally abolished in the ΔPM mutant. Considering the rif-orf19 gene was involved in the naphthalenic ring formation during rifamycin polyketide assembly, rif-orf19 was reintroduced together with rifR (type II thioesterase gene, deletion reduces metabolite production) into the ΔPM mutant to afford the ΔPM::rif-orf19+rifR strain [9]. After culturing on ISP2 agar media for 7 days at 28 °C, the fermentation products of ΔPM::rif-orf19+rifR displayed dissimilar metabolic characteristics from that of the wild-type strain through HPLC and LC-ESI-HRMS analysis. Large scale fermentation (20 L) was conducted, the fermented agar cakes were sliced and extracted. Systematic separation of the extract resulted in the identification of compounds 1–11.
The molecular formula of 1 was confirmed to be C36H47NO9 on the basis of the HRESIMS data at m/z 638.3328 [M + H]+ ion (calcd for C36H48NO9+, 638.3329) (Fig. S7). Comparison of the NMR data of 1 with that of proansamycin B (11) (Tables 1, 3, S1 and S11) indicated that 1 was 6-O-methyl proansamycin B, which was confirmed by the HMBC correlations from H-6’ (δH 3.70) to C-6 (δC 160.7).
Analysis of the HRESIMS data of compounds 2, 3 and 4 revealed that they had the same molecular formula of C35H45NO10 (Figs. S14, S20 and S27), and one more oxygen atom than that of proansamycin B. Interpretation of the NMR data determined 2, 3 and 4 to be 30-hydroxy, 31-hydroxy and 20-hydroxyproansamycin B, respectively, which was supported by the presence of C-30 as a hydroxymethyl group (δH 4.23, 4.36, δC 63.4) (Tables 1, 3 and S2), C-31 as a hydroxymethine (δH 3.43, δC 62.3) (Tables 1, 3 and S3) and C-20 as a hydroxy-substitued quaternary carbon (δC 76.4) (Tables 3 and S4).
The molecular formula of 5 was designated to be C35H43NO9 on the basis of the HRESIMS data at m/z 622.3007 [M + H]+ ion (calcd for C35H44NO9+, 622.3016) (Fig. S34). The NMR comparison with that of proansamycin B (11) (Tables 1, 3, S5 and S11) confirmed that 5 was 23-ketoproansamycin B, which was supported by the presence of C-22 (δC 48.1), C-23 (δC 219.1) and C-24 (δC 48.5) (Table 3).
Compounds 6 and 7 had the identical molecular formula of C35H43NO10 based on their HRESIMS data (Figs. S41 and S48). The NMR data revealed that both had one more oxygen atom than that of 5. The NMR comparison determined 6 to be 30-hydroxy-23-ketoproansamycin B on the basis of the presence of C-30 (δH 4.23, 4.39, δC 63.1) and C-23 (δC 219.1) (Tables 2, 3 and S6), and 7 to be 31-hydroxy-23-ketoproansamycin B on the basis of C-31 (δH 3.59, δC 64.8) and C-23 (δC 220.4) (Tables 2, 3 and S7).
The HRESIMS data at m/z 620.2867 [M + H − H2O]+ ion suggested the molecular formula of 8 to be C35H43NO10 (calcd for C35H42NO9+, 620.2965) (Fig. S55). The existence of a naphthaquinone chromophore was indicated by the HMBC correlations from H-3 (δH 7.41) to C-1 (δC 179.2), C-2 (δC 141.8), C-9 (δC 131.1) and C-10 (δC 126.0), from H-8 (δH 7.86) to C-1, C-4 (δC 185.4), C-6 (δC 158.7), C-9 and C-14 (δC 15.6), and from H-14 (δH 2.22) to C-6, C-7 (δC 131.4) and C-8 (δC 130.2) (Fig. 2 and Tables 2, 3 and S8). The existence of a twenty four-carbon fragment was determined on the basis of the 1H-1H COSY correlations and the HMBC correlations from the H-13 (δH 1.85), H-30 (δH 1.91), H-31 (δH 2.78, 3.13), H-32 (δH 0.97), H-33 (δH 0.89), H-34 (δH 0.41) and H-34a (δH 0.95) to the corresponding carbons. The hydroxylation of C-31 and oxidation of C-23 to a carbonyl group followed by the hemiketalization with the C-31 hydroxy group was confirmed on the basis of the HMBC correlations from H-31 to C-23 (δC 102.9) (Table 3, Fig. 2). The connectivity between C-5 and C-11 and the amide bond linkage between C-2 and C-15 was deduced on the basis of biosynthetic logic together with the HRESIMS data. Therefore, with the aid of biosynthetic logic and optical rotation comparisons, the structure of 8 was identified [16, 17].

Selected HMBC (⇢) and 1H-1H COSY (▬) correlations of 8, 9 and 10
Compound 9 had the molecular formula C35H41NO9 on the basis of the HRESIMS data m/z 602.2748 [M + H − H2O]+ ion (calad for C35H40NO8+, 602.2860) and 642.2674 [M + Na]+ (calcd for C35H41NO9Na+, 642.2679) (Fig. S62). A naphthaquinone chromophore was established by the HMBC correlations from H-3 (δH 6.06) to C-1 (δC 179.1), C-2 (δC 140.7), C-4 (δC 184.5) and C-9 (δC 132.7), from H-8 (δH 7.95) to C-1, C-6 (δC 158.6), C-9 and C-14 (δC 15.4), and from H-14 (δH 2.33) to C-6, C-7 (δC 131.5) and C-8 (δC 130.3) (Tables 2, 3 and S9, Fig. 2). The HMBC correlations from the H-13 (δH 1.89), H-30 (δH 2.06), H-31 (δH 1.21), H-32 (δH 1.19), H-33 (δH 0.73), H-34 (0.52), H-34a (δH 0.79) to the corresponding carbons and 1H-1H COSY correlations (Fig. 2 and Table S9) indicated the presence of a twenty four-carbon fragment. The connectivity between C-5 (δC 121.9) and C-11 and the amide bond linkage between C-2 and C-15 was deduced on the basis of biosynthetic logic together with the HRESIMS data. The C-23 hydroxymethine was oxidized to carbonyl that formed a hemiketal with the C-20 hydroxy group was revealed on the basis of the 13C NMR of C-23 (δC 112.2) (Table 3, Fig. 2). Therefore, the structure of 9 was determined.
The molecular formula of 10 was elucidated to be C35H49NO10 on the basis of the HRESIMS data m/z 644.3430 [M + H]+ ion (calcd for C35H50NO10+, 644.3435) (Fig. S69). Interpretation the NMR data of 10 exhibited the presence of a dihydronaphthoquinone chromophore in consideration of the presence of the extra protons H-2 (δH 5.20) and H-3 (δH 3.25) (Table 2). That the ansa chain occurred a retro-Claisen cleavage between C-5 and C-11 was revealed on the basis of the 13C NMR of C-11 (δC 170.6) and the extra aromatic proton H-5 (δH 7.34) compared to that of normal rifamycins, which was confirmed by the HMBC correlations from H-5 to C-4 (δC 194.2) and C-7 (δC 132.9) (Tables 3 and S10). A twenty four-carbon fragment was established on the basis of the 1H-1H COSY, and HMBC correlations from the H-13 (δH 1.85), H-30 (δH 2.00), H-31 (δH 1.02), H-32 (δH 0.94), H-33 (δH 0.96), H-34 (δH 0.80), H-34a (δH 0.95) to the corresponding carbons (Fig. 2). The amide bond linkage between C-2 (δC 53.8) and C-15 was deduced according to the intense correlations of HMBC from the H-2 to C-15. Based on the biosynthetic logic, the NOESY correlations (Fig. S68) and the optical rotation comparison with that of proansamycin B-M1 [17], the stereochemistry of aliphatic carbons of 10 was considered to be the same as that of proansamycin B (11).
The antimicrobial activity of compounds 1–11 was assayed against Pseudomonas aeruginosa PA01, methicillin-resistant Staphylococcus aureus (MRSA), Proteusbacillus vulgaris CPCC 160013 and Mycobacterium smegmatis mc2 155, respectively. The results indicated that only 1, 2 and 9 exhibited inhibitory activity against MRSA. These compounds were further measured by the microbroth dilution method, giving the MIC values of 64, 8 and 128 µg/mL for 1, 2 and 9, respectively (Table S12).
The cytotoxicity of compounds 1–11 was assayed by CCK-8 method (Bimake, USA) while adriamycin was used as the positive control. Only compound 1 exhibited significant inhibitory activity against the proliferation of MDA-MB-231, HepG2 and Panc-1 cells (Table 4). After exposure to 1 (2.5 μM) for 24 h, the percentage of annexin V-positive MDA-MB-231 cells was increased (46.04%). Flow cytometry analysis indicated a dose-dependent manner (Figs. S70 and S71).
The isolation of 1–11 from the A. mediterranei S699ΔPM::rifR+rif-orf19 mutant indicated that Rif-Orf19 was sufficient for the formation of the naphthalenic ring with the aid of non-enzymatic oxidation of hydroquinone and the followed Michael addition (Fig. 3). This result was significant in that our previous inactivation of the rif-orf19 gene in A. mediterranei S699 led to disrupting the production of rifamycins and their anticipated benzenic precursors, which was discrepant to the isolation of benzenic precursors of neoansamycins from the deletion mutant of the nam7 gene, a homolog of the rif-orf19 gene [18]. All compounds featured without C-8 hydroxy group substitution, supporting the previous conclusion that proansamycin B was an intermediate of the 8-deoxy variant shunt pathway of rifamycin B biosynthesis. The presence of C-34a methyl group indicated that its hydroxylation was specifically catalyzed by an enzyme encoded by a gene in the rif cluster rather than by a house-keeping oxidase (Fig. 3) [19]. Compounds 8 and 9 featured with a six-membered ring and a five-membered ring hemiketal, respectively, showing the involvement of non-enzymatic conversion in derivatizing rifamycins. Compound 10 experienced C-5/C-11 retro-Claisen cleavage during the biosynthesis process as exemplified by that of proansamycin B-M1 [17] and protorifamycin I–M1 [20], hygrocins I and J [21], divergolides R and S [22] and microansamycins G–I [23]. Compared with proansamycin B, compounds 2–7 illustrated diverse oxygenations at different carbons including C-20, C-23, C-30 and C-31, indicating that the ansa chain of rifamycins are easily oxidized at these specific sites (Fig. 3).

Proposed biosynthetic pathway of compounds isolated from the A. mediterranei S699ΔPM::rifR+rif-orf19 mutant
Conclusion
In the present study, the fermentation products of the mutant strain ΔPM::rifR+rif-orf19 were systematically isolated. The main product proansamycin B (11) and its ten new derivatives were obtained. Some of them had good antibacterial activity and cytotoxicity. The complex post-PKS modifications of rifamycins play a key role in enriching structural diversity and enhancing bioactivity [24]. In addition to the reported enzyme-mediated modifications, there are also a large number of non-specific modifications that are masked by the rifamycin B synthesis pathway, endowing rifamycins further structure diversity and biological activity.
Experimental section
Strains and plasmids
The strain used in the work was Amycolatopsis mediterranei S699, which was firstly separated in 1957 at St. Raphael, France, as a gift from Heinz G. Floss laboratory at University of Washington, Seattle. Firstly, the gene fragment of rif-orf3 to rif-orf16 was disturbed through homologous recombination and insertion into the shuttle plasmid pOJ260 of the E. coli-Micromonospora (Fig. S74) and the mutant of Δorf3-orf16 was selected by apramycin resistance. Secondly, the fragment of rif-orf33-rifT-rif-orf0 was removed on the basis of Δorf3-orf16 mutant according to the above method to obtain ΔPM mutant. To avoid the influence of polarity effects, the promotor rifKp was replenishmented to the gene deleted region during the construction of ΔPM mutant (Fig. S72). We verified ΔPM mutant using polymerase chain reaction (PCR) method (Fig. S75) along with a pair of primers on the genome and plasmid (Table S13). Finally, rifR and rif-orf19 genes were transformed into ΔPM mutant with electroporation technology to build a novel strain of A. mediterranei S699 ΔPM::rifR+rif-orf19. These strains need to grow on YMG (yeast extract 4 g, malt extract 10 g, glucose 4 g, 20 g agar, double distilled H2O 1000 mL, pH 7.2) agar media and be inverted in an incubator at 28 °C for product productions.
Escherichia coli DH5α strain was leveraged to conduct plasmid reproduction. E. coli cells were growing in Luria-Bertani (LB) medium (tryptone 10 g, yeast extract 5 g, NaCl 10 g, ddH2O 1000 ml, pH 7.2) and in an incubator at 37 °C. A final concentration of 50 μg/mL antibiotic apramycin was added to the LB medium of all cultivated strains. Mixing of mycelium with 20% glycerol and retained at –80 °C.
Throughout the entire study, the suicide vector pOJ260 (containing aac (3) IV, oriT, repPUC, lacZ) was used to inactivate genes within the framework. The gene expression in the ΔPM::rifR+rif-orf19 strain was completed through the integration vector pSET152 (containing aac (3) IV, oriT (RK2), ori (pUC18), int (φC31), attP (φC31), lacZα).
DNA manipulation
Compete the gene knock-out plasmid according to the following steps. Firstly, we used the A. mediterranei S699 genomic DNA as a template and amplified the upstream and downstream homologous arms (approximately 2 kb) of the target genes through PCR. Inserting the purified PCR fragments into linearized vector pOJ260 through Gibson assembly. Then chemically transformed the assembled product into 100 μL DH5α competent cells and identification of positive clones through resistance screening and DNA sequencing (Fig. S72). We used the same method as Ding et al. to delete Target genes. Plasmid was reproduced in DH5α and converted into A. mediterranei S699 competent cells through electroporation technology. Screening the recombinants produced by homologous recombination of knock-out plasmid and A. mediterranei S699 genomic DNA, transferring it into apramycin-free YMG agar for several rounds of non-selective growth (Fig. S73). Gene knockout validation was performed using PCR to screen recombinants of apramycin-sensitive from double-crossover recombination (Fig. S74). Please refer to the Supplementary Information for the specific process.
The integrating vector pSET152 was served for gene complementation in the strain of A. mediterranei S699 ΔPM::rif-orf19+rifR. The production of pSET152-rifKp was achieved by inserting the rifK promoter fragment and synthesized after digestion by NdeI and XbaI into the pSET152 vector pretreated by XbaI. Using A. mediterranei S699 genomic DNA as a template, both rif-orf19 and rifR genes were amplified by PCR. The PCR fragments of NdeI/EcoRI rif-orf19 and rifR were inserted downstream of the rifKp promoter in pSET152, respectively. The identification of positive clones were completed through PCR screening and DNA sequencing. Then, rifKp-rif-orf19 fragment was amplified by PCR using pSET152-rifKp-rif-orf19 as a template and inserted into linearized pSET152-rifKp-rifR downstream by EcoRI. The final strain of A. mediterranei S699 ΔPM::rifR+orf19 was obtained by electroporation of the resultant plasmid pSET152-rifKp-rifR-rifKp-rif-orf19 into A. mediterranei S699 ΔPM competent cells.
HPLC detection and analysis of the metabolites in mutant strains
The A. mediterranei S699 ΔPM::rifR+rif-orf19 strain was inoculated on YMG agar petri dishes and grew for 7 days in 28 °C incubator. The diced culture was extracted at room temperature overnight with EtOAc/MeOH (4 : 1, v/v). A small amount of concentrated crude extract was dissolved in an appropriate volume of methanol and analyzed through HPLC. The HPLC solvents included (A) ddH2O + 0.5% formic acid and (B) acetonitrile, UV detection was monitored at 254 nm and the flow rate was 1 mL/min. First 5 min were eluted with 20% A to 35% B gradient, the 5–15 min were eluted with 35% A to 55% B gradient, the 15–19 min were eluted with 55% A to 65% B gradient, the 19–23 min were eluted with 65% A to 100% B gradient, and finally eluted with a 100% B gradient for 4 min (Fig. S76 and S77).
General experimental procedures
NMR spectra were measured using Bruker 400 MHz and/or AVANCE 600 MHz NMR spectrometers. Column chromatography (CC) was performed on reversed-phase (RP) C18 silica gel (Merck, Darmstadt, Germany). Semi-preparative HPLC was performed on Agilent 1200 (Agilent Eclipse XDB-C18, 5 μm, 9.4 × 250 mm). Optical rotations were measured with the instrument of Auton Paar MCP200 Automatic Polarimeter. HRESIMS data were obtained with the instrument of LTQ-Orbitrap XL (Thermo Scientific, Waltham, MA, USA).
Fermentation, extraction and isolation of the metabolites in mutants
The A. mediterraneiS699 ΔPM::rifR+rif-orf19 mutant was inoculated onto 20 L YMG agar plates and grew for 7 days in 28 °C incubator. The diced culture of 20 L was extracted at room temperature with EtOAc/MeOH (4 : 1, v/v) three times overnight. The crude extract was partitioned between EtOAc and H2O (1 : 1, v/v) until the EtOAc layer was almost colorless. The organic extract was separated on RP C18 silica gel (130 g) by medium-pressure liquid chromatography (MPLC). Gradient elution was performed with 500 mL of 30%, 50%, 70%, and 100% CH3CN to obtain five fractions (Frs.), A–E. The fractions were further separated by semi-preparative HPLC, the flow rate for HPLC was 4 mL/min and the UV absorption was monitored at 254 nm.
Compound 3 (tR 15.0 min, 9.5 mg) was obtained by semi-preparative HPLC from Fr. A (0.11 g) under the condition of 28% CH3CN elution. Fr. B (0.12 g) was purified by semi-preparative HPLC with 40% CH3CN to give 7 (tR 8.2 min, 12.0 mg). Fr. C (0.39 g) was separated by semi-preparative HPLC with 42% CH3CN to afford 2 (tR 7.2 min, 6.2 mg), 6 (tR 8.9 min, 9.4 mg), 4 (tR 11.1 min, 10.5 mg), 8 (tR 13.8 min, 10.2 mg) and 10 (tR 18.6 min, 10.3 mg), respectively. Fr. D (0.42 g) was separated by semi-preparative HPLC with 45% CH3CN to obtain 11 (tR 7.8 min, 36.7 mg), 5 (tR 11.4 min, 6.6 mg) and 1 (tR 12.9 min, 9.8 mg), respectively. Compound 9 (tR 17.5 min, 16.7 mg) was purified from Fr. E (0.28 g) by semi-preparative HPLC with 52% CH3CN.
Compound 1: yellow powder; [α]20D + 60.1 (c 0.1, MeOH); UV (MeOH) λmax (logε) 229 (3.00), 266 (2.70), 300 (1.54) nm; for 1H and 13C NMR data, see Tables 1 and 3; HRESIMS: m/z 638.3328 [M + H]+ (calcd for C36H48NO9+, 638.3324).
Compound 2: tawny powder; [α]20D = +34.7 (c 0.10, MeOH); UV (MeOH) λmax (logε) 215 (3.04), 215 (2.96), 274 (2.88) nm; for 1H and 13C NMR data, see Tables 1 and 3; HRESIMS: m/z 640.3116 [M + H]+ (calcd for C35H46NO10+, 640.3116).
Compound 3: dark brown powder; [α]20D = +135.5 (c 0.10, MeOH); UV (MeOH) λmax (logε) 228 (2.76), 271 (2.13), 309 (1.12)nm; for 1H and 13C NMR data, see Tables 1 and 3; HRESIMS: m/z 640.3125 [M + H]+ (calcd for C35H46NO10+, 640.3116).
Compound 4: tawny powder; [α]20D = +98.1 (c 0.10, MeOH); UV (MeOH) λmax (logε) 230 (2.49), 273 (2.13), 309 (1.12) nm; for 1H and 13C NMR data, see Tables 1 and 3; HRESIMS: m/z 662.2938 [M + Na]+ (calcd for C35H45NO10Na+, 662.2936).
Compound 5: yellow powder; [α]20D = +175.5 (c 0.10, MeOH); UV (MeOH) λmax (logε) 228 (2.80), 267 (2.07), 309 (1.12) nm; for 1H and 13C NMR data, see Tables 1 and 3; HRESIMS: m/z 622.3007 [M + H]+ (calcd for C35H44NO9+, 622.3011).
Compound 6: tawny powder; [α]20D = +162.2 (c 0.10, MeOH); UV (MeOH) λmax (logε) 215 (3.62), 231 (3.67), 271 (3.32) nm; for 1H and 13C NMR data, see Table 2 and 3; HRESIMS: m/z 638.2970 [M + H]+ (calcd for C35H44NO10+, 638.2960).
Compound 7: tawny powder; [α]20D = +94.1 (c 0.10, MeOH); UV (MeOH) λmax (logε) 227 (3.88), 309 (2.15) nm; for 1H and 13C NMR data, see Tables 2 and 3; HRESIMS: m/z 638.2955 [M + H]+ (calcd for C35H44NO10+, 638.2960).
Compound 8: yellow powder; [α]20D = –226.9 (c 0.10, MeOH); UV (MeOH) λmax (logε) 223 (3.99), 311 (2.21) nm; for 1H and 13C NMR data, see Tables 2 and 3; HRESIMS: m/z 620.2867 [M + H − H2O] (calcd for C35H42NO9, 620.2965).
Compound 9: yellow powder; [α]20D = –172.9 (c 0.10, MeOH); UV (MeOH) λmax (logε) 219 (2.91), 310 (0.69) nm; for 1H and 13C NMR data, see Tables 2 and 3; HRESIMS: m/z 602.2748 [M + H − H2O] (calcd for C35H40NO8, 602.2860), and 642.2674 [M + Na]+ (calcd for C35H41NO9Na+, 642.2674).
Compound 10: yellow powder; [α]20D = +33.4 (c 0.10, MeOH); UV (MeOH) λmax (logε) 249 (2.61) nm; for 1H and 13C NMR data, see Tables 2 and 3; HRESIMS: m/z 628.3484 [M + H]+ (calcd for C35H50NO10+, 628.3480).
Antimicrobial assay
Compounds 1–11 against methicillin-resistant Staphylococcus aureus (MRSA) were assayed by paper disc diffusion assay in Shandong Second Provincial General Hospital. The positive control was vancomycin. The tested compounds (40 μg each) wer immersed onto the paper disks (Ø6 mm) that were then placed on the agar plates inoculated with MRSA. The test plates were incubated for 24 h at 37 °C and checked for the formation of bacteriostatic circles. The minimal inhibitory concentration (MIC) values of active compounds on MRSA growth were measured using the microbroth dilution method. Bacteria were grown in the LB media and the optical density measured at 600 nm was approximately 0.2. Equipped with 100 μL flat-bottomed microdilution trays for serial diluted compounds were inoculated into 100 μL bacteria suspensions, ultimately forming 1.0 × 103 colony forming units (CFU)/mL (spectrophotometer at 600 nm) of an inoculum size.
Cytotoxicity assay
The in vitro cytotoxicity was measured on four cell lines: L02, MDA-MB-231, HepG2, and Panc-1, respectively. All cell lines were purchased from Cell Bank of the Institute of Biochemistry and Cell Biology, China Academy of Sciences (Shanghai, China), and cultured under the standard conditions. According to the manufacturer’s instructions, cell-grown inhibition was assayed by Cell Counting Kit-8 (CCK-8) (Bimake, USA). Using adriamycin as the positive control, approximately 7 × 103 per well were seeded in 96-well plates and treated with different concentrations of compounds 1–11 for 48 h, and 10 µL of CCK-8 was added to each well and incubated for 4 h. The absorbance was measured through Spark 30086376 (TECAN, Austria), and IC50 values were calculated using software Prism 7 (GraphPad Software, Inc., San Diego, CA, USA).
References
Floss HG, Yu TW. Rifamycin-mode of action, resistance, and biosynthesis. Chem Rev. 2005;105:621–32.
Kupchan SM, Komoda Y, Court WA, Thomas GJ, Smith RM, Karim A, et al. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J Am Chem Soc. 1972;94:1354–6.
Sasaki K, Rinehart KL Jr, Slomp G, Grostic MF, Olson EC. Geldanamycin. I. structure assignment. J Am Chem Soc. 1970;92:7591–3.
Kang Q, Shen Y, Bai L. Biosynthesis of 3,5-AHBA-derived natural products. Nat Prod Rep. 2012;29:243–63.
Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, et al. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell. 2001;104:901–12.
Artsimovitch I, Vassylyeva MN, Svetlov D, Svetlov V, Perederina A, Igarashi N, et al. Allosteric modulation of the RNA polymerase catalytic reaction is an essential component of transcription control by rifamycins. Cell. 2005;122:351–63.
Ramaswamy S, Musser JM. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis. 1998;79:3–29.
Siu GK, Zhang Y, Lau TC, Lau RW, Ho PL, Yew WW, et al. Mutations outside the rifampicin resistance-determining region associated with rifampicin resistance in Mycobacterium tuberculosis. J Antimicrob Chemothe. 2011;66:730–3.
Ye F, Zhao X, Shi Y, Hu Y, Ding Y, Lu C, et al. Deciphering the timing of naphthalenic ring formation in the Biosynthesis of 8-Deoxyrifamycins. Org Lett. 2023;25:6474–78.
Han TY, Zhang K, Tang GL, Zhou Q. Characterizing Post‐PKS Modifications of 16‐Demethyl‐rifamycin revealed two dehydrogenases diverting the aromatization mode of naphthalenic ring in ansamycin biosynthesis. Chin J Chem. 2022;10:9–15.
Ye F, Shi Y, Zhao S, Li Z, Wang H, Lu C, et al. 8-Deoxy-Rifamycin Derivatives from Amycolatopsis mediterranei S699 Delta rifT Strain. Biomolecules. 2020;10:1265.
Shi YR, Ye F, Song YL, Zhang XC, Lu CH, Shen YM. Rifamycin W Analogues from Amycolatopsis mediterranei S699 Delta rif-orf5 Strain. Biomolecules. 2021;11:920.
Xu J, Wan E, Kim CJ, Floss HG, Mahmud T. Identification of tailoring genes involved in the modification of the polyketide backbone of rifamycin B by Amycolatopsis mediterranei S699. Microbioloogy. 2005;151:2515–28.
Qi F, Lei C, Li F, Zhang X, Wang J, Zhang W, et al. Deciphering the late steps of rifamycin biosynthesis. Nat Commun. 2018;9:2342.
Stratmann A, Schupp T, Toupet C, Schilling W, Oberer L, Traber R. New insights into rifamycin B biosynthesis: isolation of proansamycin B and 34a-deoxy-rifamycin W as early macrocyclic intermediates indicating two separated biosynthetic pathways. J Antibiot. 2002;55:396–406.
Hutchinson CR, Floss HG. Biosynthesis of the ansamycin antibiotic rifamycin: deductions from the molecular analysis of the rif biosynthetic gene cluster of Amycolatopsis mediterranei S699. Chem Biol. 1998;5:69–79.
Ghisalba O, Traxler P, Fuhrer H, Richter WJ. Early intermediates in the biosynthesis of ansamycins. II. Isolation and identification of proansamycin B-M1 and protorifamycin I-M1. J Antibiot. 1979;32:1267–72.
Zhang J, Li S, Wu X, Guo Z, Lu C, Shen Y. Nam7 hydroxylase is responsible for the formation of the naphthalenic ring in the biosynthesis of neoansamycins. Org Lett. 2017;19:2442–45.
Zhou Q, Luo G-C, Zhang H, Tang G-L. 34a-Hydroxylation in Rifamycin biosynthesis catalyzed by cytochrome P450 encoded by rif-orf13. Chin J Org Chem. 2019;39:58–63.
Shi Y, Zhang J, Tian X, Wu X, Li T, Lu C, et al. Isolation of 11,12-seco-Rifamycin W Derivatives Reveals a Cleavage Pattern of the Rifamycin Ansa Chain. Org Lett. 2019;21:900–03.
Li S, Lu C, Ou J, Deng J, Shen Y. Overexpression of hgc1 increases the production and diversity of hygrocins in Streptomyces sp. LZ35. RSC Adv. 2015;5:83843–46.
Zhao GS, Li SR, Guo ZX, Sun MW, Lu CH. Overexpression of div8 increases the production and diversity of divergolides in Streptomyces sp W112. Rsc Adv. 2015;5:98209–14.
Wang J, Li W, Wang H, Lu C. Pentaketide Ansamycin Microansamycins A-I from Micromonospora sp. Org Lett. 2018;20:1058–61.
Xiao YS, Zhang B, Zhang M, Guo ZK, Deng XZ, Shi J, et al. Rifamorpholines A–E, potential antibiotics from locust-associated actinobacteria Amycolatopsis sp. Hca4. Org Biomol Chem. 2017;15:3909–16.
Funding
This research was supported by the National Key Research and Development Program (2019YFA0905402), the National Natural Science Foundation of China (81673317, 81602979, 81530091), the Program for Changjiang Scholars and Innovative Research Team in University (IRT_17R68).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ma, X., Ye, F., Zhang, X. et al. Proansamycin B derivatives from the post-PKS modification gene deletion mutant of Amycolatopsis mediterranei S699. J Antibiot 77, 278–287 (2024). https://doi.org/10.1038/s41429-024-00708-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-024-00708-4
