
Abstract
We herein describe an asymmetric α-allylic allenylation of β-ketocarbonyls and aldehydes with 1,3-enynes. A synergistic chiral primary amine/Pd catalyst was identified to facilitate the utilization of 1,3-enynes as atom-economic and achiral allene precursors. The synergistic catalysis enables the construction of all-carbon quaternary centers-tethered allenes bearing non-adjacent 1,3-axial central stereogenic centers in high level of diastereo- and enantio-selectivity. By switching the configurations of ligands and aminocatalysts, diastereodivergence can be achieved and any of the four diastereoisomers can be accessed in high diastereo- and enantio- selectivity.
Similar content being viewed by others

Introduction
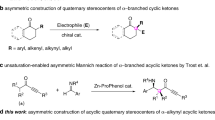
Enantioselective α-allylic allenylation of carbonyls is a powerful and straightforward approach to access chiral allenes that are of significant relevance as synthons and bio-active products1,2,3,4,5,6,7,8. Comparing with the typical allylic alkylation counterparts, allenylation process poses an additional stereochemical issue in controlling the axial chirality of allene beyond the stereogenic center construction9,10,11,12,13,14,15,16,17,18,19,20,21. A more challenging stereochemical situation is the concurrent formation of non-adjacent 1,3-axial central stereogenic centers in the α-allenylation of carbonyls (Fig. 1I)22,23,24. Progresses along this line are rather limited. Recently, enantioselective α-allylic allenylation of α-hydoxyl ketones and imine esters have been achieved with racemic allenylic esters by the work of Trost, Ma and Zhang, respectively22,24. The group of He used 1,3-enynes as atom-economic and achiral allene precursors for asymmetric allylic allenylation of α-fluoroketones23. Despite of these advances, the reactions are limited to those functionalized esters or ketones bearing additional coordinating groups. The reactions with β-ketocarbonyls and aldehydes remains underdeveloped and there is no example in the formation of all-carbon quaternary centers for this type of allylic allenylation process.

I Previous strategies on asymmetric α-Allylic allenylation of carbonyls. II Synergist amine/Pd catalysis for allenylation of ketones and aldehydes presented in this work.
Previously, we have shown metal-hydride (M-H) cycle could work in concert with chiral primary aminocatalysis to facilitate enantioselective alkylation with allene, 1,3-dienes and alkynes25,26,27,28,29,30. In this work, we report synergistic Pd/chiral primary amine catalysis for α-allylic allenylation of β-ketocarbonyls and aldehydes with the most atom-economic and achiral allene precursors, 1,3-enynes (Fig. 1II). Distinctive from the dual metal synergistic catalysis, the current organo-metal dual catalytic system eliminates the necessity on coordinating groups, and thus significantly expands the scopes to include versatile β-ketocarbonyls and aldehydes for the first time. The reaction also features the construction of all-carbon quaternary centers-tethered allenes bearing a non-adjacent 1,3-axial-central chirality, a challenging target that has not been achieved before.
Results and discussion
Reaction optimizations
Based on our preliminary successes on achiral Pd/chiral primary amine catalysis25,26,27,28,29,30, conjugated enyne 1a and racemic tert-butyl 2-methyl-3-oxobutanoate 2a were selected as the model substrates to examine the feasibility of this design in the presence of chiral primary-tertiary (S)-A1 amine catalyst, Pd2(dba)3 and DPEphos in the DCE at 40 °C (Table 1). A set of chiral primary-tertiary diamine catalysts were first examined, and A1 was found to provide 3a with 90% ee, although the yield was only 51% (entry 2). Other bulkier catalysts such as A2 slightly increased the enantioselectivity but with compromised activity (entry 3 vs entry 2). A survey of classic biphosphine ligands revealed that both of the reactivity and stereoselectivity were significantly influenced by the dihedral angle of the ligand backbone. Chiral ligand L4 with the smallest dihedral angle was identified as an efficient ligand in terms of both reactivity and stereoselectivity (entries 4–6), whereas the more sterically hindered ligand L5 gave the desired adduct with >99% ee, albeit with only trace product. Ligands L6 and L7, which were effective in other metal-hydride (M-H) mediated reactions31,32,33, turned out to be inactive in our Pd/aminocatalytic system (entries 10 and 11). Match-mismatch effect was noted on the ligand configuration. The use of (R)-L4/(S)-A1 led to a reversed diastereoselectivity, with unfortunately reduced activity and enantioselectivity (entry 6 vs entry 7). In addition, (race)-L4/(S)-A1 showed only slightly diminished stereoselectivity, suggesting that the chirality of ligand has only marginal effect on enantioselectivity and that aminocatalyst plays a major role in stereocontrol (Table 1, entry 8 vs entry 6). Further optimization identified NaBArF4 (ArF = 3, 5-(CF3)2Ph) as a promoting additive (Table 1, entry 12) and ethyl acetate (EA) as the selection of solvent for high productivity (Table 1, entries 13–17). When changing Pd2(dba)3 to Pd(PPh3)4, the desired product was obtained with high diastereoselectivity (Table 1, entry 18). Under these conditions, the swap of strong acid additive TfOH with NHTf2 further increased the yield to 74%, meanwhile maintaining >20:1 dr and >99% ee (Table 1, entry 19).
With the optimized conditions in hand, we explored the scopes of enynes by carrying out reactions with 2a, and the results are summarized in Fig. 2. A diverse array of electron-withdrawing and electron-donating groups at varying positions on the aryl units were found to be well tolerated to afford the allene products (Fig. 2, 3a–o) in 51–84% yield with excellent stereoselectivities (13:1 to >20:1 dr and exclusively >99% ee). Particularly, the enyne 1i with bromo-substitution gave the expected product 3i in 63% yield with 13:1 dr and >99% ee without obvious side products associated with the bromo-substitution in the presence of Pd. Multi-substituted arenes were also suitable as well to deliver the corresponding products in good yields with good diastereoselectivities and excellent enantioselectivities (Fig. 2, 3o, p). Heteroaryl enynes including those highly conjugate arenes were compatible with this process, providing the allene products (Fig. 2, 3q–s) in 57-76% yield with 9:1 to >20:1 dr and up to >99% ee. In addition, alkyl-substituted enyne also showed moderate coupling reactivity with >20:1 dr and >99% ee (Fig. 2, 3t).

Reaction conditions, unless otherwise noted: 1 (0.1 mmol), 2a (0.3 mmol), Pd(PPh3)4 (5 mol %), L4 (5 mol %), A1 (20 mol %), and NaBArF4 (6 mol %) in EA (0.3 mL) at 40 °C for 24 h.
The practicality of this method was examined in the late-stage allenylation of structurally complexed substrates bearing diverse biologically active molecules. For example, the reactions worked smoothly with enynes tethered to drugs such as Naproxen (3aa), (1S)-(-)-Camphanic acid (3bb), Febuxostat (3cc) and Cholesterol (3dd), to give the desired adducts with moderate reactivity and high diastereo- and enantioselectivities (Fig. 2).
We also examined the scope of β-ketocarbonyls. As show in Fig. 3A, different ester groups with varied sizes were generally tolerated to give the desired products in good yields and excellent stereoselectivities (Fig. 3A, 4a–d). Large α-substituent led to reduced reactivity and stereoselectivity (Fig. 3A, 4f), while small one worked well to give the desired adduct (Fig. 3A, 4e) in 11:1 dr, >99% ee and 64% yield. The reaction with cyclic β-ketoesters such as cyclopentanone and cyclohexanone worked well to deliver the corresponding products with excellent stereoselectivities but in lower yields (Fig. 3A and 4g–i), most likely due to the lower reactivity of these ketones.

Reaction conditions, unless otherwise noted: 1 l (0.1 mmol), 2 (0.3 mmol), Pd(PPh3)4 (5 mol %), L4 (5 mol %), A1 (20 mol %), and NaBArF4 (6 mol %) in EA (0.3 mL) at 40 °C for 24 h.
Next, the scope of β-ketoamides was evaluated for this process (Fig. 3B). For acylic β-ketoamides, the yields were decreased slightly but with significantly higher dr comparing with cyclic β-ketoamide (Fig. 3B, 4k–l vs 4m–p). To our delight, cyclic β-ketoamides turned to be favorable substrates for the reactions, showing higher activity than their β-ketoester counterparts (Fig. 3B, 4n–p vs 4h–i). Different amide groups were tolerated to give good yields and high stereoselectivities (Fig. 3B, 4j, 4n–p).
We then explored the reactions with α-branched aldehydes (Fig. 3C), for which there has been no examples on asymmetric allylic allenylation. α-Arylpropanals can be well applied and the reactions proceeded smoothly to give the desired adducts with good yields and high stereoselectivity (Fig. 3C, 4q–t). The diastereoselectivity was relatively lower comparing with that of the reactions with β-ketocarbonyls.
Synthetic applications
Stereodivergent catalysis has recently become a powerful strategy to access any stereoisomers of chiral molecules bearing multi-stereogenic centers in a highly controlled manner34,35,36,37,38,39,40,41,42,43,44,45,46,47. Early successes along this line have been achieved with chiral aminocatalysts35. In our cases, the initially identified bulky aminocatalyst A1 showed no divergence on diastereoselectivity and the switch to other configured chiral ligand resulted in only poor diastereoselectivity (Table 1 entry 7). After extensive investigation, we found the less bulky catalyst A0 showed promising results (Fig. 4) and the combination of A0 and L4/L2 with varied configurations led to any of the four diastereoisomers in good yields, high enantioselectivities and with moderate to good diastereoselectivities (up to 67% yield, 15:1 dr, and >99% ee).

Stereodivergent synthesis of all the four stereoisomers of 3 h by the combination of A0 and L4/L2 with varied configurations.
The reaction between 1l and 2a could be conducted at a gram scale with comparable results (Fig. 2, 3l), demonstrating the practicality of the current reaction. In addition, this series of products are rich in functionality for further transformations that were enormously demonstrated48,49,50,51,52. In our cases, the initially obtained allylic allene adducts could be easily transformed in order to determine the stereochemical configurations. Upon treatment of 3a with 2, 4-dinitrophenylhydrazine followed by HCl (1 M, 1.0 equiv.) in MeOH led to compound 5a in 67% yield and 19: 1 dr with 99% ee (Fig. 5, 5a). Reduction of the keto moiety of (S,S)-3a over NaBH4 gave 5b in 98% yield (Fig. 5,5b). The absolute configuration of reduction product 5b was assigned based on crystallographic analysis.

Reaction conditions: a 2,4-dinitrophenylhydrazine (1.2 equiv.), 1 M HCl (1.0 equiv.), MeOH (0.1 M), r.t. b NaBH4, MeOH (0.5 M), r.t., 2 h.
Proposed catalytic cycle
In accordance with previous studies23,25,26,27,28,29,30, a synergistic Pd/aminocatalytic cycle could be proposed as described in Fig. 6. For palladium catalysis, a L*/Pd-H complex generated in situ underwent hydrocarbonation with enyne regioselectively to give the intermediate I, which could interconvert rapidly to the key π-allyl-palladium syn-η3-int II via σ-π rearrangement15,53,54. The protonated aminocatalyst may serve as the proton source for Pd-H formation. Subsequently, intermediate syn-η3-Int II coupled with the enamine intermediate III to give the desired product 3a after hydrolysis. On the basis of experimental observations as well as our previous studies27, a steric model could be proposed to account for the observed stereoselectivity regarding the chiral center construction (Fig. 6, III), whereas chiral ligand in synergy with aminocatalysts plays the key role in steering the axial selectivity.

Synergistic catalytic cycle of aminocatalysis and palladium catalysis.
In summary, we have developed a synergistic chiral primary amine/Pd catalysis for α-allylic allenylation of ketones and aldehydes. The catalysis enables the use of 1,3-enynes as the atom-economic and achiral allene precursors, and features high stereoselectivity in the construction of non-adjacent 1,3-axial central chirality bearing all-carbon quaternary centers. This method could be generally applied to β-ketocarbonyls and aldehydes to afford the allenylation products with high enantioselectivity and diastereoselectivity. Divergence on the stereoselectivity could be achieved by varying the configurations of chiral primary amine and chiral ligands.
Methods
General procedure
In glove box, to a flame-dried Schlenk tube equipped with a magnetic stir bar was added tert-butyl 2-methyl-3-oxobutanoate (2a, 0.30 mmol), but-3-en-1-yn-1-ylbenzene (1a, 0.10 mmol), Pd(PPh3)4 (5 mol%), (S)-SEGphos (5 mol%), NaBArF4 (6 mol%) and primary amine (S)-A1 (20 mol%), the mixture was diluted with 0.3 mL of anhydrous EA, then the mixture was moved out from glove box and stirred at 40 °C for 24 h, solvent was removed and residue was purified by silica gel chromatography (10% EtOAc in Petroleum ether) to give 3a as a colorless oil. The enantiometric excess was determined by HPLC (AD-H*2).
Data availability
All data are available from the authors upon request. Supplementary information and chemical compound information are available along with the online version of the paper. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition number 2222345 (5b). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
References
Krause, N. & Hashmi, A. S. K. Modern Allene Chemistry (Wiley-VCH, Weinheim, 2004).
Ma, S.-M. Some typical advances in the synthetic applications of allenes. Chem. Rev. 105, 2829–2872 (2005).
Ye, J.-T. & Ma, S.-M. Palladium-catalyzed cyclization reactions of allenes in the presence of unsaturated carbon-carbon bonds. Acc. Chem. Res. 47, 989–1000 (2014).
Yu, S.-C. & Ma, S.-M. How easy are the syntheses of allenes? Chem. Commun. 47, 5384–5418 (2011).
Neff, R. K. & Frantz, D. E. Recent advances in the catalytic syntheses of allenes: a critical assessment. ACS Catal. 4, 519–528 (2014).
Ye, J.-T. & Ma, S.-M. Conquering three-carbon axial chirality of allenes. Org. Chem. Front. 1, 1210–1224 (2014).
Huang, X. & Ma, S.-M. Allenation of terminal alkynes with aldehydes and ketones. Acc. Chem. Res. 52, 1301–1312 (2019).
Liu, Y. & Bandini, M. B. Nickel catalyzed functionalization of allenes. Chin. J. Chem. 37, 431–441 (2019).
Imada, Y., Ueno, K., Kutsuwa, K. & Murahashi, S. I. Palladium-catalyzed asymmetric alkylation of 2,3-alkadienyl phosphates. synthesis of optically active 2-(2,3-alkadienyl)malonates. Chem. Lett. 31, 140–141 (2002).
Trost, B. M., Fandrick, D. R. & Dinh, D. C. Dynamic kinetic asymmetric allylic alkylations of allenes. J. Am. Chem. Soc. 127, 14186–14187 (2005).
Nemoto, T., Kanematsu, M., Tamura, S. & Hamada, Y. Palladium-catalyzed asymmetric allylic alkylation of 2,3-allenyl acetates using a chiral diaminophosphine oxide. Adv. Synth. Catal. 351, 1773–1778 (2009).
Zhang, W. et al. Enantioselective bromolactonization of conjugated (Z)-enynes. J. Am. Chem. Soc. 132, 3664–3665 (2010).
Hashimoto, T., Sakata, K., Tamakuni, F., Dutton, M. J. & Maruoka, K. Phase-transfer-catalysed asymmetric synthesis of tetrasubstituted allenes. Nat. Chem. 5, 240–244 (2013).
Mbofana, C. T. & Miller, S. J. Diastereo- and enantioselective addition of anilide-functionalized allenoates to N-acylimines catalyzed by a pyridylalanine-based peptide. J. Am. Chem. Soc. 136, 3285–3292 (2014).
Song, S.-H., Zhou, J., Fu, C.-L. & Ma, S.-M. Catalytic enantioselective construction of axial chirality in 1,3-disubstituted allenes. Nat. Commun. 10, 507 (2019).
Wu, G.-L. et al. Enantioselective allenation of terminal alkynes catalyzed by copper halides of mixed oxidation states and its application to the total synthesis of scorodonin. Angew. Chem. Int. Ed. 61, e202112427 (2022).
Zhang, F.-H., Guo, X.-C., Zeng, X.-R. & Wang, Z.-B. Catalytic enantioconvergent allenylation of aldehydes with propargyl halides. Angew. Chem. Int. Ed. 61, e202117114 (2022).
Yu, X.-Z., Ren, H.-J., Xiao, Y.-J. & Zhang, J.-L. Efficient assembly of allenes, 1,3-dienes, and 4H-pyrans by catalytic regioselective nucleophilic addition to electron-deficient 1,3-conjugated enynes. Chem. Eur. J. 14, 8481–8848 (2008).
Yao, Q. et al. Efficient synthesis of chiral trisubstituted 1,2-allenyl ketones by catalytic asymmetric conjugate addition of malonic esters to enynes. Angew. Chem., Int. Ed. 55, 1859–1863 (2016).
Dai, J.-X., Duan, X.-Y., Zhou, J., Fu, C.-L. & Ma, S. M. Catalytic enantioselective simultaneous control of axial chirality and central chirality in allenes. Chin. J. Chem. 36, 387–391 (2018).
Poulsen, P. H. et al. Organocatalytic formation of chiral trisubstituted allenes and chiral furan derivatives. Angew. Chem. Int. Ed. 57, 10661–10665 (2018).
Trost, B. M., Schultz, J. E., Chang, T. & Maduabum, M. R. Chemo-, regio-, diastereo-, and enantiose palladium allylic alkylation of 1,3-dioxaboroles as synthetic equivalents of α-hydroxyketones. J. Am. Chem. Soc. 141, 9521–9526 (2019).
Yang, S.-Q., Wang, Y. F., Zhao, W.-C., Lin, G.-Q. & He, Z.-T. Stereodivergent synthesis of tertiary fluoride-tethered allenes via copper and palladium dual catalysis. J. Am. Chem. Soc. 143, 7285–7291 (2021).
Zhang, J.-C. et al. Enantio- and diastereodivergent construction of 1,3-nonadjacent stereocenters bearing axial and central chirality through synergistic Pd/Cu catalysis. J. Am. Chem. Soc. 143, 12622–12632 (2021).
Zhou, H., Zhang, L., Xu, C.-M. & Luo, S.-Z. Chiral primary amine/palladium dual catalysis for asymmetric allylic alkylation of β-ketocarbonyl compounds with allylic alcohols. Angew. Chem. Int. Ed. 54, 12645–12648 (2015).
Zhou, H., Wang, Y.-N., Zhang, L., Cai, M. & Luo, S.-Z. Enantioselective terminal addition to allenes by dual chiral primary amine/palladium catalysis. J. Am. Chem. Soc. 139, 3631–3634 (2017).
Wang, Y.-N. et al. Steric Effect of protonated tertiary amine in primary-tertiary diamine catalysis: a double-layered sterimol model. Org. Lett. 21, 407–411 (2019).
Wang, Y.-N. et al. π-Coordinating chiral primary amine/palladium synergistic catalysis for asymmetric allylic alkylation. J. Am. Chem. Soc. 142, 3184–3195 (2020).
Wang, Y.-N., Zhang, J., You, C., Mi, X.-L. & Luo, S.-Z. Catalytic asymmetric addition and telomerization of butadiene with enamine intermediates. CCS Chem. 4, 2267–2275 (2022).
Zhang, J. et al. Asymmetric coupling of β-ketocarbonyls and alkynes by chiral primary amine/Rh synergistic catalysis. Org. Lett. 24, 1186–1189 (2022).
Kim, J. & Cho, S. H. Access to enantioenriched benzylic 1,1-silylboronate esters by palladium-catalyzed enantiotopic-group selective suzuki-miyaura coupling of (diborylmethyl)silanes with aryl iodides. ACS Catal. 9, 230–235 (2019).
Zhang, Q.-L. et al. Stereodivergent coupling of 1,3-dienes with aldimine esters enabled by synergistic Pd and Cu catalysis. J. Am. Chem. Soc. 141, 14554–14559 (2019).
Mu, D.-L. et al. Enantioselective synthesis of acyclic monohydrosilanes by steric hindrance assisted C-H silylation. Chem. Commun. 58, 7388 (2022).
Le, T. P., Tanaka, S., Yoshimura, M., Sato, K. & Kitamura, M. Stereodivergent dehydrative allylation of β-keto esters using a Ru/Pd synergistic catalyst. Nat. Commun. 13, 5876 (2022).
Krautwald, S., Sarlah, D., Schafroth, M. A. & Carreira, E. M. Enantio- and diastereodivergent dual catalysis: α-allylation of branched aldehydes. Science 340, 1065–1068 (2013).
Shi, S.-L., Wong, Z.-L. & Buchwald, S. L. Copper-catalysed enantioselective stereodivergent synthesis of amino alcohols. Nature 532, 353–356 (2016).
Trost, B. M., Hung, C. I., Saget, T. & Gnanamani, E. Branched aldehydes as linchpins for the enantioselective and stereodivergent synthesis of 1,3-aminoalcohols featuring a quaternary stereocentre. Nat. Catal. 1, 523–530 (2018).
Liu, X.-J. et al. Sequence-dependent stereodivergent allylic alkylation/fluorination of acyclic ketones. Angew. Chem. Int. Ed. 59, 2039–2043 (2020).
Changotra, A., Bhaskararao, B., Hadad, C. M. & Sunoj, R. B. Insights on absolute and relative stereocontrol in stereodivergent cooperative catalysis. J. Am. Chem. Soc. 142, 9612–9624 (2020).
Zhu, D.-X., Liu, J.-G. & Xu, M. H. Stereodivergent synthesis of enantioenriched 2,3-disubstituted dihydrobenzofurans via a one-pot C-H functionalization/oxa-michael addition cascade. J. Am. Chem. Soc. 143, 8583–8589 (2021).
Huo, X.-H. et al. Stereodivergent Pd/Cu catalysis for asymmetric desymmetric alkylation of allylic geminal dicarboxylates. CCS Chem. 4, 1720–1731 (2022).
Peng, Y.-B., Huo, X.-H., Luo, Y.-C., Wu, L. & Zhang, W.-B. Enantio- and diastereodivergent synthesis of spirocycles through dual-metal-catalyzed [3+2] annulation of 2-vinyloxiranes with nucleophilic dipoles. Angew. Chem. Int. Ed. 60, 24941–24949 (2021).
Xiao, L. et al. Cooperative catalyst-enabled regio- and stereodivergent synthesis of α-quaternary α-Amino acids via asymmetric allylic alkylation of aldimine esters with racemic allylic alcohols. Angew. Chem. Int. Ed. https://doi.org/10.1002/anie.202212948 (2022).
Wang, H.-F., Zhang, R.-Y., Zhang, Q.-L. & Zi, W.-W. Synergistic Pd/amine-catalyzed stereodivergent hydroalkylation of 1,3-dienes with aldehydes: reaction development, mechanism, and stereochemical origins. J. Am. Chem. Soc. 143, 10948–10962 (2021).
Han, J.-Q., Liu, R.-X., Lin, Z.-T. & Zi, W.-W. Stereodivergent construction of Csp3−Csp3 bonds bearing vicinal stereocenters by synergistic palladium and phase-transfer catalysis. Angew. Chem. Int. Ed. 62, e202215714 (2022).
Zhang, Q.-L., Zhu, M.-H. & Zi, W.-W. Synergizing palladium with Lewis base catalysis for stereodivergent coupling of 1,3-dienes with pentafluorophenyl acetates. Chem 8, 2784–2796 (2022).
Zhu, M.-H., Wang, P.-X., Zhang, Q.-L., Tang, W.-J. & Zi, W.-W. Diastereodivergent aldol-type coupling of alkoxyallenes with pentafluorophenyl esters enabled by synergistic palladium/chiral lewis base catalysis. Angew. Chem. Int. Ed. 61, e202207621 (2022).
Ma, S.-M. & Gao, W.-Z. Efficient synthesis of 4-(2’-alkenyl)−2,5-dihydrofurans and 5,6-dihydro-2H-pyrans via the Pd-catalyzed cyclizative coupling reaction of 2,3- or 3,4-allenols with allylic halides. J. Org. Chem. 67, 6104–6112 (2022).
Ma, S.-M., Yu, F. & Gao, W.-Z. Studies on Pd(II)-catalyzed coupling-cyclization of α- or β-amino allenes with allylic halides. J. Org. Chem. 68, 5943–5949 (2003).
Jiang, X.-P., Fu, C.-L. & Ma, S.-M. Highly stereoselective iodolactonization of 4,5-allenoic acids-an efficient synthesis of 5-(1’-iodo-1’(Z)-alkenyl)−4,5-dihydro-2(3H)-furanones. Chemistry 14, 9656–9664 (2008).
Campolo, D., Gastaldi, S., Roussel, C. & Bertrand, M. P. Axial-to-central chirality transfer in cyclization processes. Chem. Soc. Rev. 42, 8434–8466 (2013).
Neff, R. K. & Frantz, D. E. Recent applications of chiral allenes in axial-to-central chirality transfer reactions. Tetrahedron 71, 7–18 (2015).
Ogasawara, M. et al. Synthesis, structure, and reactivity of (1,2,3-η3-butadien-3-yl)palladium complexes. Organometallics 26, 5025–5029 (2007).
Wan, B.-Q. & Ma, S.-M. Enantioselective decarboxylative amination: synthesis of axially chiral allenyl amines. Angew. Chem. Int. Ed. 52, 441–445 (2013).
Acknowledgements
We thank the Natural Science Foundation of China (21861132003 and 22031006 to S.L.) and Tsinghua University Initiative Scientific Research Program (to S.L.) for financial support.
Author information
Authors and Affiliations
Contributions
S.L. and X.M. conceived and directed the project. C.Y. optimized the reaction conditions, examined the substrate scope and studied the mechanism with the help of S.M. C.Y. and S. L. wrote the manuscript with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Weiwei Zi, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
You, C., Shi, M., Mi, X. et al. Asymmetric α-allylic allenylation of β-ketocarbonyls and aldehydes by synergistic Pd/chiral primary amine catalysis. Nat Commun 14, 2911 (2023). https://doi.org/10.1038/s41467-023-38488-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-38488-4
This article is cited by
-
Catalytic stereodivergent allylic alkylation of 2-acylimidazoles for natural product synthesis
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
