
Abstract
To enable high performance of all solid-state batteries, a catholyte should demonstrate high ionic conductivity, good compressibility and oxidative stability. Here, a LaCl3-based Na+ superionic conductor (Na1−xZrxLa1−xCl4) with high ionic conductivity of 2.9 × 10−4 S cm−1 (30 °C), good compressibility and high oxidative potential (3.80 V vs. Na2Sn) is prepared via solid state reaction combining mechanochemical method. X-ray diffraction reveals a hexagonal structure (P63/m) of Na1−xZrxLa1−xCl4, with Na+ ions forming a one-dimensional diffusion channel along the c-axis. First-principle calculations combining with X-ray absorption fine structure characterization etc. reveal that the ionic conductivity of Na1−xZrxLa1−xCl4 is mainly determined by the size of Na+-channels and the Na+/La3+ mixing in the one-dimensional diffusion channels. When applied as a catholyte, the NaCrO2||Na0.7Zr0.3La0.7Cl4||Na3PS4||Na2Sn all-solid-state batteries demonstrate an initial capacity of 114 mA h g−1 and 88% retention after 70 cycles at 0.3 C. In addition, a high capacity of 94 mA h g−1 can be maintained at 1 C current density.
Similar content being viewed by others

Introduction
Rechargeable sodium-ion all-solid-state batteries (ASSBs) have attracted much attention in recent years because of their safety and cost advantages over conventional lithium-ion batteries with liquid electrolytes1,2,3,4. Sodium solid-state electrolytes (SSEs) are one of the critical components in sodium-ion ASSBs, dictating the electrochemical performance of ASSBs. To ensure high power density, high-energy density, long cycle life, and low cost of ASSB, SSE should exhibit high ionic conductivity in a wide temperature range, broad electrochemical window, excellent compatibility with electrode materials, and low cost. Various types of sodium SSEs have been developed for their applications in sodium ASSBs, including organic polymers1, inorganic sulfides5,6,7,8,9,10,11,12, oxides13,14,15,16, halides4,17,18,19,20,21,22,23, and borohydrides24,25,26.
Polymer-based SSEs are cheap and easy to process, but the ionic conductivity cannot match the industry requirement1. Oxides demonstrate ionic conductivity up to 10−3 S cm−1, but they are incompatible with cathode materials (e.g., Na3V2(PO4)3), leading to enormous interface resistance between the SSE and the cathode13,14,27. Sulfides show the highest ionic conductivity of 10−2 S cm−1, but its inferior electrochemical stability results in poor cycle stability when coupling with Na metal anodes and layered structure cathodes3,12,28. Borohydrides also have high ionic conductivity, but the low oxidation potential is unstable against high-potential cathodes24,26. In comparison, halides have been considered as a promising SSE because of their high ionic conductivity, great deformability, adequate chemical stability, and good oxidative stability against cathodes. For example, plenty of lithium-ion halide SSEs with high ionic conductivity up to 10−2 S cm−1 have been developed, such as Li3YCl6, Li3InCl6, Li3ScCl6, Li2ZrCl6, and LiTaOCl4 et al.29,30,31,32,33,34,35.
Several sodium halide SSEs have also been developed recently, such as Na2ZrCl6, Na3YCl6, Na3ErCl6, NaAlCl4, and their derivatives4,18,21,22,23. Although these sodium halides exhibit oxidation stability as good as lithium halides, their sodium ionic conductivity is much lower (~10−5 S cm−1 at room temperature). Thus, it is urgent to develop sodium halide SSEs with high ionic conductivity. Typical halide electrolyte is composed of MX6 (M = Y3+, Zr4+, Sc3+ et al. cations, X = F−, Cl−, Br−, I− et al. halides) octahedron frame and alkali metal ions (Li+, Na+, et al.) are distributed in the interstitial sites. Compared to lithium SSEs, the charge carrier of Na+ in sodium SSEs is much larger than Li+ (106 pm vs. 76 pm), which requires a significantly larger diffusion channel to ensure fast Na+ ion diffusion.
Broaden the ion diffusion channel by adopting anions or cations with larger sizes is a promising strategy to increase the ionic conductivity of SSEs. For example, NASICON is a typical SSE structure, which can achieve high Li+/Na+ ion conductivity up to 10−3 S cm−1 with optimized compositions, such as Li1+xAlxGe2−x(PO4)3, Li1+xAlxTi2−x(PO4)3 and Na3Zr2Si2PO1213,14,16,36,37,38. In Li-NASICON, the transition metal ions are mainly Ge4+ and Ti4+, with ionic radius of 53 pm and 60.5 pm. While in Na-NASICON, the transition metal changes to Zr4+, which has a bigger radius of 72 pm compared to Ge4+ and Ti4+. Li10GeP2S12 and Na11Sn2PS12 are both LISICON type ionic conductors with high ionic conductivity (10−3 ~ 10−2 S/cm), here Sn4+ (69 pm) in Na-LISICON is also bigger than Ge4+ (53 pm) in Li-LISICON39,40. Therefore, selecting larger cations, such as La3+ (103 pm), Pr3+ (99 pm), and Sm3+ (96 pm), could potentially increase the Na+ ionic conductivity of sodium halide SSEs.
Recently, a class of MCl3 (M = La-Gd) based halide SSEs has been reported, in which the Li+ ion conductivity can reach 10−3 S/cm41,42. In this structure, MCl9 tricapped trigonal prisms are stacked along the c-axis, enclosing one-dimensional channels. Li+ ions hop between two neighbored octahedral sites with a short distance of 2.08 Å in this one-dimensional diffusion channel. Similarly, the octahedral sites can also be occupied by Na+ ions, as Lissner et al. reported in 1993 (Na3xM2−xCl6, M = La-Sm)43. As shown in Fig. 1a, Na+ ions partly occupy the octahedral sites along the c-axis, with a slightly larger distance of 2.19 Å between neighbored sites, making Na3xM2−xCl6 (M = La-Sm) a potential Na+-ion superionic conductor.

Crystal structure of the NaLaCl4 (the white ball: vacancy) (a). Rietveld refinements of NaLaCl4-HM (b), Na0.9La0.9Zr0.1Cl4-HM (c), Na0.8La0.8Zr0.2Cl4-HM (d), Na0.7La0.7Zr0.3Cl4-HM (e) and lattice parameters of Na1−xLa1−xZrxCl4-HM obtained from XRD refinement (f).
In this work, we prepared and optimized the Na+ ion diffusion channel in NaLaCl4 (Na0.5(Na0.25La0.75)Cl3) with Zr4+ doping (Na1−xLa1−xZrxCl4 or Na0.5−0.75x(Na0.25La0.75−0.75xZr0.75x)Cl3), which achieved a high ionic conductivity of 2.9 × 10−4 S cm−1 (30 °C). Detailed structure characterizations and theoretical simulations indicate that the doping of Zr4+ expands the sodium-ion migration pathway, thereby promoting fast ion conduction along the c axis. In addition, the oxidation stability of Na1−xLa1−xZrxCl4 is on par with other types of halide SSEs. The ASSB using Na1−xLa1−xZrxCl4 as catholyte shows excellent electrochemical performance, which can retain about 94 mA h g−1 capacity at 1 C current and 88% capacity after 70 cycles at 0.3 C.
Result and discussion
Synthesis and characterizations of Na1−xLa1−xZrxCl4
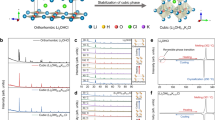
High-temperature sintered NaLaCl4 (NLC-HT) exhibits the same structure as LaCl3 (P63/m), and the synthesized material did not contain detectable impurities (Supplementary Fig. 1). This material can be considered as Na+ doped LaCl3 (Na0.5(La0.75Na0.25)Cl3), in which 25% of La3+ in the 2c site is replaced by Na+, and the rest Na+ takes the interstitial site 2b. Rietveld refinement (Supplementary Fig. 2a and Supplementary Table 1) indicates the lattice parameters of a = b = 7.567 Å and c = 4.346 Å. The enriched Na+ vacancies at 2b sites and the short distance (2.19 Å) between adjacent sites could potentially benefit the rapid migration of Na+. However, the ionic conductivity of NLC-HT is as low as 10−9 S cm−1, and the migration activation energy is as large as 0.65 eV. The main reason for the low ionic conductivity of NaLaCl4 could come from the large migration barrier for Na+. In other words, the migration path for Na+ ion diffusion is too narrow. Thus, we adopted Zr4+ doping at the La3+ site (2c) to broaden the diffusion channel. In addition, the higher valence state of Zr4+ compared to La3+ can introduce more Na+ vacancies in the structure that will be beneficial to its ionic conductivity.
As shown in Supplementary Figs. 1 and 2, the maximum Zr4+ doping amount in NaLaCl4 is around 0.2 (Na0.8Zr0.2La0.8Cl4) without observing significant impurity phases. The a lattice parameter increases from 7.567 Å to 7.573 and 7.578, and then decreases to 7.545 Å with the increase of Zr4+ content, The c lattice parameter exhibits similar trends that increases from 4.346 Å to 4.353 Å and 4.357 Å, and then decreases to 4.359 Å (Supplementary Fig. 3, Supplementary Tables 2–4). The lattice parameters increase with the amount of dopant until impurity phases appear at the Zr4+ doping level of 0.3, suggesting the successful doping of Zr4+ in NaLaCl4. The increase lattice parameters by Zr4+ doping decreased the Na+ migration energy to 0.36 eV (NLZC0.2-HT, Supplementary Figs. 4 and 5) that is comparable to other Na+ SSEs18. In addition, the ionic conductivity is significantly improved from 10−9 S cm−1 (NLC-HT) to 10−6 S cm−1 (NLZC0.2-HT) although further improvement is still needed for its practical application.
To further promote the ionic conductivity, the high-temperature sintered samples were ball milled to introduce disordering and defects, which can further increase the doping concentration of Zr4+ to expand Na+ diffusion channel (NLZCx-HM). As shown in Supplementary Fig. 1b, the impurities in NLZC0.3-HT disappear after ball milling (NLZC0.3-HM) that can be attributed to the improved thermodynamic stability of Zr-doped NaLaCl4 with enriched defects and higher entropy through ball milling. However, when the doping level of Zr4+ is increased to 0.4 (NLZC0.4-HM), the impurities cannot be fully removed even after ball milling treatment. Compared to high-temperature sintered samples (NLZCx-HT), the lattice parameter a in NLZCx-HM changes to 7.565 Å (x = 0), 7.570 Å (x = 0.1), 7.570 Å (x = 0.2) and 7.605 Å (x = 0.3) after ball milling, while lattice parameter c increases to 4.372 Å (x = 0), 4.372 Å (x = 0.1), 4.372 Å (x = 0.2) and 4.400 Å (x = 0.3).
The nonlinear change in lattice parameters implies the occurrence of other defects induced by the ball milling process, such as anti-site defects. X-ray diffraction (XRD) refinement results in Fig. 1b–e and Supplementary Tables 5–8 reveal that part of La3+ would take the 2b site and further expands the lattice. Synchrotron-based XRD of NaLaCl4-HT and NaLaCl4-HM were carried out to uncover the changes in fine structure and the reason for volume expansion. The refinement results shown in Supplementary Fig. 6 and Supplementary Table 9 reveal a small amount of Na+ at the 2b site (0.16) in NaLaCl4-HT, which means this site is metastable for Na+ occupying. Excess Na exists in the form of unknown impurities (Supplementary Fig. 6). During the ball milling process, the high energy leads to more Na+ occupancy at the 2b site (0.251), resulting in the disappearance of impurities and lattice expansion (Supplementary Fig. 7 and Supplementary Table 10). Moreover, the expansion of the lattice stabilizes the Na+ at the 2b site. Zr4+ doping at La3+ site can also stabilize Na+ at 2b site and result in more Na+ occupancy at the 2b site (0.461) in Na0.7La0.7Zr0.3Cl4-HT (Supplementary Fig. 8 and Supplementary Table 11), leading to an increase of lattice parameter a (7.583 Å). No significant crystal structural changes were found in the framework, such as the decrease in orderliness (Supplementary Figs. 6 and 7), which could be related to the strong bond energy of M-Cl that helps keep its crystal structure.
Ion conduction in Na1 − xLa1−xZrxCl4
The structural change induced by ball milling and the expanded lattice is beneficial for the Na+ ion conduction in Na1−xZrxLa1−xCl4. As a result, the ionic conductivity is further increased by one to two orders of magnitude (Fig. 2a, b and Supplementary Fig. 9). Especially for the NLZC0.3-HM sample, its ionic conductivity reaches 2.9 × 10−4 S cm−1 that is much higher than other sodium halide SSEs reported in literature4,18,22,23,44. Moreover, its activation energy reduces to 0.33 eV, lower than the same type SSEs (Fig. 2c). In addition, the electronic conductivity of NLZC0.3-HM is measured to be 1.3 × 10−8 S cm−1 (Fig. 2d), which is at the similar level compared to other types of SSEs28.

a Arrhenius conductivity plots of NLZCx-HM (0≤x ≤ 0.4), b ionic conductivities and activation energies of NLZCx-HM (0 ≤ x ≤ 0.4), c ionic conductivity and activation energy comparing with Na2ZrCl6 (A)18, Na2.4Er0.4Zr0.6Cl6 (B)44, NaAlCl4 (C)22, Na2.25Y0.25Zr0.75Cl6 (D)4 from literature, d DC polarization curve of Na0.7La0.7Zr0.3Cl4-HM under 0.5 V.
The significantly increased ionic conductivity with Zr doping (Na1−xZrxLa1−xCl4) was studied through theoretical simulations and X-ray absorption fine structure (XAFS). There are abundant channels available for one-dimensional (1D) ion diffusion in rare earth UCl3 (U = La-Sm) halides (Supplementary Fig. 10a) with the P63/m lattice. When this 1D channel is occupied by alkali metal ions, such as Li+ (Li3xLa2−xCl6), Na+ (Na3xLa2−xCl6 in Supplementary Fig. 10b), they may quickly migrate along this one-dimensional channel. Here, rapid Na+ ions diffusion can be achieved in NLZC by doping Zr4+ into NaLaCl4 (Supplementary Fig. 10c). By randomly generating 2 × 2 × 3 supercell structures and conducting structural relaxation (Fig. 3a), the most likely NLZC structures (Fig. 3b) were selected based on the criteria of energy and unit cell distortion. With the doping of Zr4+ at the La3+ site, the higher valence and smaller size of Zr4+ (0.89 Å vs. 1.216 Å for La3+) leads to shorter M-Cl bond (2.64 Å for Zr and 2.96 Å for La). As a result, the bond length of NaCl in the 1D channel increases from the original 2.87 Å to 2.91 Å (Fig. 3c). The longer NaCl bond broadens the diffusion bottleneck in the 1D channel, lowers the site energy of Na+ ions at the bottleneck, thereby lowering their migration activation energy and increasing the Na+ ion conductivity. The AIMD simulations imply that the migration of Na+ ions in NLZC requiring a lower barrier energy comparing to NLC (0.04 eV in NLZC vs. 0.26 eV in NLC, Fig. 3d). It has been found that the ion diffusion coefficient along the c direction is two orders of magnitude higher than along the ab plane for much lower migration barrier in previous studies41, suggesting that NLZC is a 1D conductor.

a Normalized electrostatic energy of 500 Na-free configurations in NLZC supercell model with representative high-energy (upper) and low-energy (lower), and corresponding results of the relaxation of two of the structures (Na ions were removed for clearer exhibition). b 221 supercell of NLZC for better displaying the c axis channel. c bond length for La-Cl, Zr-Cl, and NaCl in NLC or NLZC. d Arrhenius plot of Na+ migration pathways. Diffusivity in the NLZC lattice from AIMD simulations. e Na+ probability density, represented by green isosurfaces from AIMD simulations at 300 K in the NLZC lattice.
For this 1D conductor, ion mixing along the diffusion channel should have a significant impact on the Na+ ion conduction. A small amount of La3+ (0.06) occupy the 2b site along the c axis, due to the similar radius of La3+ (1.216 Å) and Na+ (1.24 Å with 9 coordinations) (Supplementary Table 8). The high valence of La3+ enhances the La-Cl bond energy and decreases its mobility, thus the La3+ on the migration channel should block the migration of Na+. Similar phenomena have also been found in other one-dimensional conductors, such as LiFePO445. Thus, the effect of Na+/La3+ mixing on sodium-ion conduction is also simulated. To save the computation time, a larger mixing ratio of 0.17 was constructed in 2 × 2 × 3 supercell. AIMD was used to study the change of 1D ionic conductivity before and after doping Zr4+. The NLZC-La/Na-mixing structure was constructed with energy as the criterion (Supplementary Fig. 11). The thermodynamic stability of the constructed model was also investigated (Supplementary Fig. 12). The comparison of results shows that when the NLZC is at 400 K, the structural frame with LaCl3 as the premise has greater distortion, which means it is more unstable, while the NLZC-La/Na-mixing still keeps the structure relatively stable under 500 K, indicating that the mixing makes the material more stable. The ionic conductivity of NLZC is 13.84 × 10−3 S cm−1 at 300 K, while that of NLC is only 1.15 × 10−9 S cm−1, which explains the improvement of ionic conductivity caused by Zr4+ doping as shown in Fig. 3d. Considering the La/Na mixing at 2b site (NLZC-La/Na mixing), the ionic conductivity decreases to 5.93 × 10−3 S cm−1 at 300 K. In addition, this atomic-mixing leads to a larger migration barrier (0.21 eV) comparing to that without atomic-mixing (0.04 eV). Moreover, the Na-ion migration of NLZC is much higher than that of NLC and NLZC-La/Na mixing through the probability density distribution (Fig. 3e and Supplementary Fig. 13) of AIMD.
The Zr K-edge and La L3-edge XAFS were obtained to reveal the local coordination structure of Na0.7La0.7Zr0.3Cl4-HT, Na0.7La0.7Zr0.3Cl4-HM and NaLaCl4-HT. It can be seen that the absorption edge of the Zr K-edge and La L3-edge of three basically coincide (Supplementary Fig. 14), indicating equal valence with Zr4+ and La3+. From the R-space of Zr K-edge, the main peak at about 2 Å could be recognized as Zr-Cl coordination from Na0.7La0.7Zr0.3Cl4-HT and Na0.7La0.7Zr0.3Cl4-HM (Supplementary Fig. 15). Similarly, the R-space curve of La L3-edge also show a significant difference in the position of the main peaks compare with La2O3 (Supplementary Fig. 16), which could be recognized as La-Cl coordination of NaLaCl4, Na0.7La0.7Zr0.3Cl4-HT and Na0.7La0.7Zr0.3Cl4-HM. As a result, Zr-Cl bond and La-Cl bond lengths are different in the same sample.
As shown in Fig. 4, EXAFS fitting was performed to quantitatively compare the Zr-Cl and La-Cl coordination structures in Na0.7La0.7Zr0.3Cl4, and the results are summarized in Supplementary Table 12. NaLaCl4, NLZC0.3-HT, and NLZC0.3-HM have a similar bond length of La-Cl and the coordination numbers of La, which proves that the replacing of the Zr element and ball milling process does not change its structure, which is consistent with the XRD results (Supplementary Fig. 1). It can be confirmed that the bond length of Zr-Cl is about 2.48 Å while it is 2.94 Å for La-Cl. These results confirm that the doping of Zr in the La site shortens the bond length of M-Cl, which broadens the diffusion path of Na+ ions along the c axis, as revealed from theoretical simulations (Fig. 3). In addition, the coordination numbers of Zr and La are close, further confirming the successes doping of Zr at the La site, although the radius of Zr4+ is significantly different from La3+.

a La-Cl coordination structures. b Zr-Cl coordination structures.
Electrochemical performance of ASSBs using NLZC0.3-HM
The typical electrochemical stable window of sodium halide SSE is from 1.5 V to 3.7 V4. Thus, sodium halide SSEs are mainly used as catholytes to match high-potential cathodes. Linear scanning voltammetry (LSV) of NLZC0.3-HM-Super P||NLZC0.3-HM||Na3PS4||Na2Sn reveals that the electrochemical stable window of NLZC0.3-HM is from 1.33 V to 3.80 V vs. Na2Sn at 30 °C and from 1.44 V to 3.79 V vs. Na2Sn at 60 °C (Supplementary Fig. 17), which is consistent with the theoretical results of sodium halide SSEs4. The high reduction potential of NLZC0.3-HM indicates that this SSE is thermodynamically unstable against Na2Sn or Na metal anodes. To verify the dynamic stability of NLZC0.3-HM against low potential anodes and its possibility as an anolyte, symmetric cell of Na2Sn||NLZC0.3-HM||Na2Sn was assembled and cycled (Supplementary Fig. 18). When cycled at current density with 0.01 mA cm−2, the voltage increases to 0.1 V within only 70 h, showing the instability of Na2Sn/NLZC0.3-HM interface (Supplementary Fig. 18). Both La 3d and Zr 3d peaks shift to the low-energy direction, indicating the reduction of Zr4+ and La3+ after cycling (Supplementary Fig. 19). Thus, this SSE cannot be directly used as an anolyte.
ASSBs were assembled with homemade NaCrO2 (Supplementary Fig. 20) composite cathode, Na2Sn anode, and NLZC0.3-HM SSE, with Na3PS4 serving as a transition layer to isolate the NLZC0.3-HM electrolyte from the Na2Sn anode (Fig. 5a). From the SEM image of the ASSB cross-section (Supplementary Fig. 21), the thickness of these four layers is determined to be ~60 μm (NaCrO2 composite cathode), ~300 μm (NLZC0.3-HM SSE), ~400 μm (Na3PS4 SSE) and ~70 μm (Na2Sn anode) respectively. Enlarged SEM image (Fig. 5b) and EDS (Supplementary Fig. 22) confirm the tight physical contact between NaCrO2 cathode and NLZC0.3-HM SSE, which should be beneficial to the Na+ ion diffusion at the interface. The two SSEs layers and Na2Sn anode layer are quite dense with only small amount pores (Fig. 5c–e). The boundaries between layers are relatively clear, with little mixing between them (Supplementary Figs. 22–25). The total resistance of these ASSBs before cycling is about 400 Ω (Supplementary Fig. 26), which is contributed by NLZC0.3-HM (~150 Ω, Supplementary Fig. 6d), Na3PS4 SSE (~80 Ω, Supplementary Fig. 27), Na2Sn anode side (~20 Ω, Supplementary Figs. 27 and 28a), resistance between NLZC0.3-HM and Na3PS4 (~50 Ω, Supplementary Fig. 28a, b), and interface from NaCrO2 cathode side (~100 Ω, Supplementary Figs. 26 and 28b). Na2.9PS3.9Cl0.1 with higher ionic conductivity (Supplementary Fig. 29) was also selected as a transition layer to reduce the total resistance of ASSB. However, this electrolyte reacts with NLZC0.3-HM, and the resistance keeps increasing when co-pressed together (Supplementary Fig. 30). The XPS spectra indicate structural changes with NLZC0.3-HM and NPSC mixing together (Supplementary Fig. 31), especially for NPSC, which causes an increase in resistance (Supplementary Fig. 30).

a Configuration of ASSB with NLZC0.3-HM as SSEs. Cross-section SEM for NaCrO2 composite (b), Na0.7La0.7Zr0.3Cl4 SSE (c), Na3PS4 SSE (d), and Na2Sn anode (e) in the ASSB. f charge-discharge profiles at different currents (0.1 C, 0.2 C, 0.3 C, 0.5 C, 1 C between 2 and 3.4 V vs. Na/Na+, 30 °C), g rate performance, h charge–discharge profiles at different cycles (0.3 C, 30 °C), i cycle performance.
The ASSB of NaCrO2||NLZC0.3-HM||Na3PS4||Na2Sn was cycled between 2.0 V and 3.4 V, in which range the NLZC0.3-HM SSE is electrochemically stable. When cycling at 0.1 C (30 °C), the discharge capacity for the first cycle reaches 123 mA h g−1, with a high Columbic efficiency (CE) of 95%, reflecting a highly irreversible reaction in this ASSB. When current density increases, the capacity decreases from 123 mA h g−1 (0.1 C) to 119 mA h g−1 (0.2 C), 114 mA h g−1 (0.3 C), 108 mA h g−1 (0.5 C), and 94 mA h g−1 (1 C), showing high-capacity retention at high current density (Fig. 5f, g). Compared to other halide SSEs with lower ionic conductivity, the rate performance of ASSB using NLZC0.3-HM is better4,46. After cycling at 0.3 C for 70 times, the capacity retains 100 mA h g−1, which is 88% of the initial capacity (Fig. 5h, i). Part of the capacity loss should come from an increase in resistance, which is 780 Ω after 70 cycles at 0.3 C (Supplementary Fig. 26). This increase in resistance may originate from Na3PS4/Na2Sn interface, and further enhancement in cycle performance can be achieved with other SSE transition layers, such as Na4(B12H12)(B10H10)46. However, this is still much better than ASSB using sulfide catholyte (10% capacity retention after only 20 cycles at 0.1 C), indicating much better stability of NLZC0.3-HM (Supplementary Fig. 32).
In summary, a sodium halide SSE of Na0.7La0.7Zr0.3Cl4 with a space group of P63/m was designed and synthesized. In this structure, La3+, Zr4+ and partly Na+ occupied a tricapped trigonal prisms site (MCl9) and stack along the c-axis, in which the one-dimensional channel is enclosed between them. The rest of Na+ ions take the sites in this one-dimensional channel and diffuse along the channels, giving high ionic conductivity. Structure characterizations and theoretical simulation results reveal that the doping of Zr4+ in the La3+ site shortens the length of the M-Cl bond (M = Zr4+, La3+), which broaden Na+ ion diffusion path and improves Na+ ion conductivity. On the other hand, a small amount of La3+ in the one-dimensional diffusion path blocks Na+ ion diffusion, which reduces the ionic conductivity. As a result, Na0.7La0.7Zr0.3Cl4 achieves a decent ionic conductivity of 2.9 × 10−4 S cm−1 (30 °C) and a low activation energy of 0.33 eV. Finally, the electrochemical stable window of Na0.7La0.7Zr0.3Cl4 is determined to be as high as 3.7 V vs. Na/Na+, enabling high stability against the NaCrO2 cathode. Promising electrochemical performance of NaCrO2/Na2Sn all-solid-state cells using Na0.7La0.7Zr0.3Cl4 as the catholyte is achieved, with high initial CE of 95% and 88% capacity retention after cycling at 0.3 C for 70 times. In addition, a high capacity of 94 mA h g−1 can be obtained at 1 C current.
Methods
Material synthesis
The raw materials of NaCl, ZrCl4, and LaCl3 were all purchased from Sinopharm without further purification. NaCl, ZrCl4, and LaCl3 were hand milled with a Na: Zr: La: Cl molar ratio of (1-x): x: (1−x): 4 for 30 min. The pre-mixed powders were then sintered at 450 °C for 10 h under an Ar environment. After that, the samples were ball-milled (QM-3SP2, Nanda Instruments) in a 50 ml ZrO2 jar, with a speed of 550 rpm for 10 h under vacuum. All operations were performed in an Ar-filled glovebox (Mikrouna, H2O < 0.1 ppm, O2 < 0.1 ppm).
Na2.9PS3.9Cl0.1 powders were prepared according to our previous reports47. A stoichiometric mixture of Na2S (Sigma Aldrich), P2S5 (99%, Sigma Aldrich), and NaCl (Sinopharm) was ball milled at 550 rpm for 2 h and sintered at 300 °C for 2 h under Ar. After ground again, the powders were sintered at 420 °C for another 12 h under Ar. Na3PS4 electrolytes were prepared via mechanochemical method. Na2S (Sigma Aldrich) and P2S5 (Sigma Aldrich) with a stoichiometric ratio were ball-milled at 500 rpm for 12 h. The mixed powders were vacuum sealed in a quartz tube and annealed at 280 °C for 1 h.
The NaCrO2 cathode was prepared via a solid-state reaction. Na2CO3 and Cr2O3 (Sinopharm) were mixed with a Na:Cr ratio of 1.05:1, 5% excess Na to compensate for the volatilization at high temperatures. The mixture was first ball milled at 550 rpm for 10 h under vacuum and then sintered at 900 °C for 10 h (OTF-1200X, HF-Kejing) under Ar. When cooling to 200 °C, the sample was transferred to the glovebox quickly to reduce sample exposure to air.
Na2Sn anode was prepared with ball milling Na and Sn metal. The stoichiometric mixture of Na and Sn metal (Sinopharm) was first rolled together and then transferred into a steel jar, sealed under Ar. The mixture was ball milled at 300 rpm for 10 h to obtain the homogenous product. If the product is not homogenous, another 10 h ball milling is needed.
Characterizations
Powder XRD experiments were performed using a Philips X’Pert powder diffractometer (D/MAX2500VL/PC, Rigaku) at 45 kV and 40 mA with Cu-Kα radiation (λ = 1.5406 Å). The samples were placed in a zero-background holder and sealed with a Kapton film to avoid air exposure. The data was collected at room temperature with 2θ from 10o to 80o.
Rietveld refinement of Na1−xZrxLa1−xCl4 were carried out from XRD data with strong intensity. The following parameters were refined step wisely: (1) scale factor, (2) background using linear interpolate function with 10 coefficients, (3) peak shape using the pseudo-Voigt function, (4) unit cell parameters and fractional atomic coordinates, (5) fractional occupancy and thermal displacement parameters (Uiso).
Synchrotron XRD experiments were carried out at XPD beamline (X-ray Powder Diffraction, ID28) at the National Synchrotron Light Source II (NSLS-II), Brookhaven National Laboratory, USA, with a photon wavelength of 0.185794 Å.
X-ray photoelectron spectroscopy was performed using a PHI 5000 VersaProbe III with a monochromatic Al Kα X-ray source.
The X-ray absorption spectra (XAS) including X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) of the sample at Zr K-edge and La L3-edge collected at Beijing Synchrotron Radiation Facility 1W1B beamline. Samples were covered with Kapton film to avoid air exposure.
The ionic conductivity of Na1−xZrxLa1−xCl4 was measured at 25 °C using Ac electrochemical impedance spectroscopy (EIS) in the frequency range from 1 MHz to 1 Hz with a potential perturbation of 50 mV (Biologic SP-200). The activation energy was calculated based on variable-temperature impedance from room temperature up to 100 °C in a microclimate chamber. The as-synthesized Na1−xZrxLa1−xCl4 powders were pressed into pellets with a diameter of 12 mm, under a pressure of 260 MPa, and then sandwiched by two steel rods for all measurements.
The electrochemical stability of Na0.7La0.7Zr0.3Cl4 was characterized with cyclic voltammetry. Na0.7La0.7Zr0.3Cl4 was mixed with 30% SP and used as the cathode, Na3PS4 was used as the SSE, and Na2Sn was used as the anode. These three parts were respectively pressed under 300 MPa in a cylinder cell with a diameter of 12 mm. The cyclic voltammetry was carried on Biologic SP-200 with a scan rate of 0.2 mV/s and voltage window of 1.3–4.9 V.
All-solid-state NaCrO2||NLZC0.3-HM||Na3PS4||Na2Sn battery was fabricated using the following procedure. The composite cathode was made by ball milling the mixture of NaCrO2, NLZC0.3-HM, and SP (50: 50: 3 in weight ratio) at 300 rpm for 30 min. The ASSB was made by co-pressing Na2Sn anode (50 mg), Na3PS4 (70 mg) anolyte, NLZC0.3-HM (70 mg) catholyte, and NaCrO2 composite cathode (12 mg) together in order, and under 300 MPa. The ASSB was cycled between 2.0 and 3.4 V vs. Na/Na+, under current densities of 0.1 C, 0.2 C, 0.3 C, 0.5 C, 1 C, respectively. The specific capacity is calculated based on the mass of NaCrO2 cathode.
SEM image of the assembled ASSBs were conducted on a higher solution field emission scanning electron microscope (Hitachi). Before that, the ASSBs were polished using a cooling cross-section polisher (JEOL) to obtain a smooth surface.
Density functional theory calculations
The first-principles calculations were conducted using the Vienna Ab initio Simulation 5.4.4 Package based on density functional theory48,49. The exchange-correlation interaction was generalized gradient approximation (GGA) using the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional while the projector augmented-wave (PAW) pseudopotential was used to account electron-ion interactions50,51,52. The cutoff energy for the plane-wave basis was set to 400 eV, and the total energy convergence was set to be lower than 1 × 10−5 eV, with the force convergence at 0.03 eV Å−1, the integrations in the reduced Brillouin zone are performed on a Γ-centered 3 × 3 × 3 special k-points for all calculations.
Ab initio molecular dynamics simulations
In order to realize the fractional occupied structures, the structure of 2 × 2 × 3 supercell of NLZC0.3 and NLC was randomly generated using supercell software package, and the lowest electrostatic potential 500 structures were output, structural convergence was carried out respectively. The optimal structure was selected based on lattice difference and energy. Considering that La/Na mixing appears in the 2c site of the synthesized materials, the mixing structure is selected based on the criteria of energy and lattice difference, mixing structure were also screened in 2 × 2 × 3 supercell for reducing the time consuming.
The AIMD simulation of the supercell model was carried out to study ion diffusion, and the non-spin polarization model was used. The SCF convergence criterion of 10−4 eV in the NVT ensemble lasted above 40 ps with a time step of 2 fs until the diffusivity (D/cm2 s−1) converges53. D is calculated as the mean azimuth shift (MSD) over the time interval Δt:
where N is the total number of diffusion ions, d is the dimension of the diffusion system, rNa(t) is the displacement of the Na-th ion at time t and the bracket represents averaging over t. Ionic conductivity (σ/S cm−1) was derived from the Nernst–Einstein relationship:
Where n is the number of diffusion ions per unit volume (Å3), z is ionic charge number, e is the electron charge (C).
Data availability
The data that support the findings of this study are available within the article (and its Supplementary Information files) and from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Wang, X., Zhang, C., Sawczyk, M. & Sun, J. Ultra-stable all-solid-state sodium metal batteries enabled by perfluoropolyether-based electrolytes. Nat. Mater. 21, 1057–1065 (2022).
Chi, X. et al. An electrochemically stable homogeneous glassy electrolyte formed at room temperature for all-solid-state sodium batteries. Nat. Commun. 13, 2854 (2022).
Hayashi, A. et al. A sodium-ion sulfide solid electrolyte with unprecedented conductivity at room temperature. Nat. Commun. 10, 5266 (2019).
Wu, E. A. et al. A stable cathode-solid electrolyte composite for high-voltage, long-cycle-life solid-state sodium-ion batteries. Nat. Commun. 12, 1256 (2021).
Hayashi, A., Noi, K., Sakuda, A. & Tatsumisago, M. Superionic glass-ceramic electrolytes for room-temperature rechargeable sodium batteries. Nat. Commun. 3, 856 (2012).
Zhu, Z., Chu, I.-H., Deng, Z. & Ong, S. P. Role of Na+ interstitials and dopants in enhancing the Na+ conductivity of the cubic Na3PS4 superionic conductor. Chem. Mater. 27, 8318–8325 (2015).
de Klerk, N. J. J. & Wagemaker, M. Diffusion mechanism of the sodium-ion solid electrolyte Na3PS4 and potential improvements of halogen doping. Chem. Mater. 28, 3122–3130 (2016).
Richards, W. D. et al. Design and synthesis of the superionic conductor Na10SnP2S12. Nat. Commun. 7, 11009 (2016).
Che, H. et al. Electrolyte design strategies and research progress for room-temperature sodium-ion batteries. Energy Environ. Sci. 10, 1075–1101 (2017).
Zhang, L. et al. Vacancy‐contained tetragonal Na3SbS4 superionic conductor. Adv. Sci. 3, 1600089 (2016).
Famprikis, T. et al. A new superionic plastic polymorph of the Na+ conductor Na3PS4. ACS Mater. Lett. 1, 641–646 (2019).
Fuchs, T., Culver, S. P., Till, P. & Zeier, W. G. Defect-mediated conductivity enhancements in Na3-xPn1-xWxS4 (Pn = P, Sb) using aliovalent substitutions. ACS Energy Lett. 5, 146–151 (2020).
Zhang, L. et al. Negating Na||Na3Zr2Si2PO12 interfacial resistance for dendrite-free and “na-less” solid-state batteries. Chem. Sci. 9, 44–51 (2018).
Deng, Z. et al. Fundamental investigations on the sodium-ion transport properties of mixed polyanion solid-state battery electrolytes. Nat. Commun. 13, 4470 (2022).
Yang, J. et al. Ultrastable all-solid-state sodium rechargeable batteries. ACS Energy Lett. 5, 2835–2841 (2020).
Ma, Q. et al. Enhancing the dendrite tolerance of NaSICON electrolytes by suppressing edge growth of Na electrode along ceramic surface. Adv. Energy Mater. 12, 2201680 (2022).
Qie, Y. et al. Yttrium–sodium halides as promising solid-state electrolytes with high ionic conductivity and stability for na-ion batteries. J. Phys. Chem. Lett. 11, 3376–3383 (2020).
Kwak, H. et al. Na2ZrCl6 enabling highly stable 3 V all-solid-state Na-ion batteries. Energy Storage Mater. 37, 47–54 (2021).
Park, D. et al. Materials design of sodium chloride solid electrolytes Na3MCl6 for all-solid-state sodium-ion batteries. J. Mater. Chem. A 9, 23037–23045 (2021).
Huang, H. et al. Fast ion transport mechanism and electrochemical stability of trivalent metal iodide-based Na superionic conductors Na3XI6 (X = Sc, Y, La, and In). ACS Appl. Mater. Interfaces 14, 36864–36874 (2022).
Li, R., Xu, K., Liu, K., Si, R. & Zhang, Z. Computational screening of Na3MBr6 compounds as sodium solid electrolytes. Chem. Mater. 34, 8356–8365 (2022).
Park, J. et al. NaAlCl4: new halide solid electrolyte for 3 V stable cost-effective all-solid-state Na-ion batteries. ACS Energy Lett. 7, 3293–3301 (2022).
Kwak, H. et al. Boosting the interfacial superionic conduction of halide solid electrolytes for all-solid-state batteries. Nat. Commun. 14, 2459 (2023).
Cuan, J. et al. Borohydride‐scaffolded Li/Na/Mg fast ionic conductors for promising solid‐state electrolytes. Adv. Mater. 31, 1803533 (2019).
Unemoto, A., Matsuo, M. & Orimo, S. Complex hydrides for electrochemical energy storage. Adv. Funct. Mater. 24, 2267–2279 (2014).
Sadikin, Y., Brighi, M., Schouwink, P. & Černý, R. Superionic conduction of sodium and lithium in anion-mixed hydroborates Na3BH4B12H12 and (Li0.7Na0.3)3BH4B12H12. Adv. Energy Mater. 5, 1501016 (2015).
Ma, Q. et al. Room temperature demonstration of a sodium superionic conductor with grain conductivity in excess of 0.01 S cm-1 and its primary applications in symmetric battery cells. J. Mater. Chem. A 7, 7766–7776 (2019).
Feng, X. et al. Heavily tungsten‐doped sodium thioantimonate solid‐state electrolytes with exceptionally low activation energy for ionic diffusion. Angew. Chem. Int. Ed. 60, 26158–26166 (2021).
Asano, T. et al. Solid halide electrolytes with high lithium-ion conductivity for application in 4 V class bulk-type all-solid-state batteries. Adv. Mater. 30, 1803075 (2018).
Wang, K. et al. A cost-effective and humidity-tolerant chloride solid electrolyte for lithium batteries. Nat. Commun. 12, 4410 (2021).
Tanaka, Y. et al. New oxyhalide solid electrolytes with high lithium ionic conductivity >10 mS/cm for all‐solid‐state batteries. Angew. Chem. Int. Ed. 62, e202217581 (2023).
Li, X. et al. Water‐mediated synthesis of a superionic halide solid electrolyte. Angew. Chem. Int. Ed. 58, 16427–16432 (2019).
Liang, J. et al. Site-occupation-tuned superionic LixScCl3+x halide solid electrolytes for all-solid-state batteries. J. Am. Chem. Soc. 142, 7012–7022 (2020).
Kwak, H. et al. New cost‐effective halide solid electrolytes for all‐solid‐state batteries: mechanochemically prepared Fe3+‐substituted Li2ZrCl6. Adv. Energy Mater. 11, 2003190 (2021).
Zhou, L. et al. A new halospinel superionic conductor for high-voltage all solid state lithium batteries. Energy Environ. Sci. 13, 2056–2063 (2020).
Mousavi, T. et al. Fabrication of Li1+xAlxGe2-x(PO4)3 thin films by sputtering for solid electrolytes. Solid State Ion. 354, 115397 (2020).
Xiao, W., Wang, J., Fan, L., Zhang, J. & Li, X. Recent advances in Li1+xAlxTi2−x(PO4)3 solid-state electrolyte for safe lithium batteries. Energy Storage Mater. 19, 379–400 (2019).
Yang, K., Chen, L., Ma, J., He, Y. & Kang, F. Progress and perspective of Li1+xAlxTi2-x(PO4)3 ceramic electrolyte in lithium batteries. InfoMat 3, 1195–1217 (2021).
Duchardt, M., Ruschewitz, U., Adams, S., Dehnen, S. & Roling, B. Vacancy-controlled Na+ superion conduction in Na11Sn2PS12. Angew. Chem. Int. Ed. 57, 1351–1355 (2018).
Kamaya, N. et al. A lithium superionic conductor. Nat. Mater. 10, 682–686 (2011).
Fu, J. et al. Superionic conducting halide frameworks enabled by interface-bonded halides. J. Am. Chem. Soc. 145, 2183–2194 (2023).
Yin, Y.-C. et al. A LaCl3-based lithium superionic conductor compatible with lithium metal. Nature 616, 77–83 (2023).
Lissner, F. et al. Die chloride Na3xM2-xC1, (M = La-Sm) und NaM2Cl6, (M = Nd, Sm): derivate des UC13-Typs. Synthese, Kristallstruktur und Rontgenabsorptionsspektroskopie (XANES). Z. Anorg. Allg. Chem. 620, 444–450 (1994).
Schlem, R., Banik, A., Eckardt, M., Zobel, M. & Zeier, W. G. Na3-xEr1-xZrxCl6—a halide-based fast sodium-ion conductor with vacancy-driven ionic transport. ACS Appl. Energy Mater. 3, 10164–10173 (2020).
Islam, M. S., Driscoll, D. J., Fisher, C. A. J. & Slater, P. R. Atomic-scale investigation of defects, dopants, and lithium transport in the LiFePO4 olivine-type battery material. Chem. Mater. 17, 5085–5092 (2005).
Ridley, P. et al. Glass-ceramic sodium-deficient chlorides with high sodium-ion conductivity. https://chemrxiv.org/engage/chemrxiv/article-details/637a89cf20798134fe2e6586 (2022).
Feng, X. et al. Studies of functional defects for fast Na‐ion conduction in Na3-yPS4-xClx with a combined experimental and computational approach. Adv. Funct. Mater. 29, 1807951 (2019).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
He, X., Zhu, Y., Epstein, A. & Mo, Y. Statistical variances of diffusional properties from ab initio molecular dynamics simulations. Npj Comput. Mater. 4, 18 (2018).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (U2330101 to X.F., 52072105 to H.X., 12305368 to W.X. and 22073087 to X.W.), the National Natural Science Foundation for Distinguished Young Scholars (22225301 to X.W.), the Anhui Provincial Natural Science Foundation (2108085J23 to H.X.), the Major Science and Technology Projects in Anhui Province (202203a05020032 to X.F., 2022e03020004 to X.F., 202003a05020014 to H.X. and 2021e03020001 to H.X.), the CAS Project for Young Scientists in Basic Research (YSBR-004 to X.W.), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0450101 to X.W.), the Fundamental Research Funds for the Central Universities (JZ2022HGTB0251 to X.F. and 20720220009 to X.W.). The authors would also thank the Beijing Synchrotron Radiation Facility 1W1B beamline for X-ray Absorption Fine Structure characterization and the Super Computer Center of USTCSCC and SCCAS. This research also used resources of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704 (A.A.). We acknowledge Dr. Nan Wang at Brookhaven National Laboratory for the help in handling samples for the synchrotron measurement.
Author information
Authors and Affiliations
Contributions
X.F. and H.X. conceived the research. C.F. and W.G. performed the materials synthesis, conductivity measurement, structural characterization, Rietveld refinement, and electrochemical tests. Y.L. and X.W. conducted theoretical simulations. W.X. completed the XAFS analysis. A.A. conducted the synchrotron XRD measurements, and L.S. and J.L. completed the analysis. X.F. wrote the manuscript under the assistance of C.F., L.Z., W.D., and W.W. X.F. and H.X. directed the entire study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fu, C., Li, Y., Xu, W. et al. LaCl3-based sodium halide solid electrolytes with high ionic conductivity for all-solid-state batteries. Nat Commun 15, 4315 (2024). https://doi.org/10.1038/s41467-024-48712-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48712-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
