
Abstract
Electrochemical separations are powerful platforms for the sustainable recovery of critical elements, environmental remediation and downstream processing. However, the recent development of electroseparations has primarily focused on heterogeneous adsorbents, which face the challenge of intermittent electroswing operation. Here we present a redox-mediated electrochemical liquid–liquid extraction separation platform that translates selective single-site binding to a fully continuous separation scheme. A redox-active extractant is molecularly designed with controllable hydrophobicity to maximize organic phase retention. The redox flow design enables fully electrified continuous operation with no external chemical input, achieving the selective recovery of precious metals from multicomponent streams. We demonstrate an atomic efficiency of over 90% and over 100:1 selectivity for practical critical metal leach streams, and 16-fold up-concentration for gold and platinum group metals from varied feedstocks including electronic waste, catalytic converter waste and mining streams. Our work is envisioned as a pathway towards a broader class of industrially applicable liquid–liquid extraction-based electrochemical separations.

Similar content being viewed by others
Main
Electrochemical separations offer a resource-efficient platform for critical metal recovery that can integrate with a renewable-energy infrastructure in a plug-and-play fashion1,2. Most notably, redox-mediated electro-sorbents have displayed remarkable molecular specificity, being previously demonstrated in the energy-efficient purification of value-added products and the removal of dilute contaminants3,4,5,6,7,8,9. However, electrode-based adsorbent systems are inherently an intermittent process, necessitating frequent electrode regeneration to release the captured target species10. Translation to a fully continuous electrochemical process could enhance their direct industrial applicability by providing ease of scalability and modularity11,12. Here we describe the development of an electrochemical liquid–liquid extraction (e-LLE) platform that combines the power of a single-site redox-mediated extractant with solvent extraction design principles to enable highly selective and continuous electrochemical separation.
Electrochemical separations have been demonstrated as a promising approach to efficient and sustainable resource capture and purification13,14. By coating electrodes with redox-active materials, increased charge capacity and selective ion capture have been achieved15,16,17,18, including as a modular platform for the energy-efficient purification of value-added materials8,19. Polyvinyl ferrocene (PVF)-functionalized electrodes have been shown to enable the recycling of precious-metal homogeneous catalysts in situ, without destruction of the active catalyst structure, enabling extended turnovers8. Electronic structure investigations have determined the underlying mechanism to be favorable charge-transfer interactions between ferrocenium (Fc+) and anionic targets, leading to the selective capture of transition-metal complexes8,16.
Immobilized redox-active ferrocene (Fc) units have shown a high affinity for ionic gold complexes, enabling gold purification and refinement with high energy efficiency19. However, these redox electrosorption schemes are intrinsically limited by the swing nature of adsorption, with the increased system complexities associated with this intermittency often hindering the economy of scale needed for large-scale critical element recovery and purification10. Although rocking-chair-type designs provide pseudo-continuous operation of electrosorption20, the operational difficulty of switching can still impact the efficiency of these schemes. Electrodialysis as an electrochemical separation platform has demonstrated commercial success owing to its continuous operation, but its viability is limited by the selectivity and cost of the required membranes21.
Liquid–liquid extraction (LLE or LLX) is a powerful and widely implemented technology in industry that has enabled efficient, scalable and fully continuous chemical separation, particularly in critical metal recovery22,23. Complex extractant chemistries have been developed to effectively capture aqueous precious-metal complexes into the organic extractant phase24,25. However, metal release from the extractant phase (stripping) is instrinsically slow and chemically intensive with traditional extractants, and relies on wasteful thermal or chemical swings. Difficulties in regenerating a target metal from the extractant have historically limited the economic viability of classical LLE and posed challenges to the sustainability of these separation processes26,27,28. Here, by creating a framework for e-LLE, our study proposes the utilization of a hydrophobic modified Fc unit as an electrochemically mediated extractant for the selective and efficient recovery of precious metals from dilute, multicomponent metal leach solutions. We design a redox extractant that can be rapidly switched on and off with a mild electrochemical potential, resulting in the complete release and up-concentration of target species for separations.
Precious metals such as gold, platinum, palladium and iridium are among the most environmentally damaging and costly elements to be mined because of the expensive separation and processing technologies required29, the high carbon footprints of their refinement, and their increasing demand and decreasing ore grade30,31,32,33,34. Gold is used extensively in electronics35 (accounting for 8% of gold demand), and current recycling processes are inefficient and environmentally polluting35,36,37, with low economic viability38,39, meaning that over 90% of the gold used in electronics is sequestered to landfills in the United States each year40. Similarly, platinum group metals (PGMs) face substantial recovery challenges due to their presence in multicomponent mixtures, as well as their low abundance and the high technological demand for catalytic materials and components critical to renewable-energy technologies41,42.
This work investigates e-LLE through the rational molecular design of a Fc redox extractant with high binding affinity towards gold and PGMs. By increasing the functionalized hydrocarbon chain length, the Fc extractant is retained to the organic extractant phase with minimal loss to the aqueous phase. The resulting 1,1′-didodecylferrocene (ddFc) was found to effectively extract over 99% of aqueous gold into organic dichloromethane (DCM), demonstrating exceptional molecular efficiency (0.9 mol Au extracted for every mole of ddFc extractant) and selectivity. Selective gold uptake and up-concentration were achieved in the e-LLE system in a fully continuous manner using practical gold leach solutions derived from local electronic waste and mining ore, with only electrical input. We also carried out the extraction and purification of PGMs from automotive catalytic converter material, and selective extraction of platinum and iridium chloro-complexes was achieved. A techno-economic analysis of the e-LLE system demonstrated a substantial lowering of the cost of the gold-recovery process, with promising applications for the recovery of low-grade gold. By simultaneously enhancing the recovery performance and reducing energy and material consumption, the e-LLE process offers a framework for the design of electrochemical separations that extends beyond membrane- and adsorption-based processes, expanding its scope into highly scalable and adaptable LLE processes for critical and precious metal recovery and purification.
Electrochemical architecture design
A schematic of our e-LLE process is shown in Fig. 1a,c. Our approach uses a redox-active Fc-based extractant designed to remain sequestered to the organic phase (shown in blue). The organic extractant stream is looped through three consecutive solvent extraction columns: oxidation, leach and reduction columns. The ddFc extractant is activated by oxidation in the first column via an aqueous oxidizing agent, shown in red. From there, the oxidized ddFc in the organic stream contacts the aqueous gold-containing leach stream (yellow) in the second extraction column, and anionic dicyanoaurate, [Au(CN)2]−, binds favorably to the oxidized ddFc extractant, selectively transferring the gold complex from the aqueous to the organic phase. The gold-barren leach stream (raffinate) exiting the e-LLE process can then be post-treated to capture any remaining base metals and dissolved organic solvent. The gold is reversibly released and concentrated into a new aqueous stream (in green) in the third and final extraction column. The dicyanoaurate anion is then liberated to the aqueous phase simply by reducing ddFc+ back to ddFc with an aqueous reducing agent, ferrocyanide. Pure metallic gold is efficiently recovered from the aqueous reducing loop (in green) via electrodeposition. The organic stream containing reduced ddFc extractant is then pumped back to the first extraction column, in a closed loop. Additionally, the oxidizing agent (in red) and reducing agent (in green) are simultaneously regenerated in a membrane-separated flow cell. The selective recovery and purification of gold is thus achieved continuously, solely based on electrical input. The proposed e-LLE system brings significant advantages over both prior electrosorption platforms and competing industrial technologies (Fig. 1b).

a, Schematic of the continuous e-LLE system, which consists of three solvent extraction columns and a membrane-separated flow cell. b, Overall comparison of the ddFc e-LLE extraction system (blue) to previous work (red) and the standard industrial technology (gray). c, A detailed reaction schematic describing the e-LLE system consisting of three solution loops. In red is the aqueous oxidation loop with iodine. In blue is the organic extractant loop with ddFc in DCM. In green is the aqueous reduction loop with ferrocyanide.
Molecular design and selection of redox-active extractant
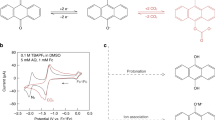
Redox electrodes have previously been demonstrated to selectively bind to anionic cyano-gold via a directly applied oxidizing potential, with reversible gold release when electrically reduced19. The Fc unit was selected as an effective organic extractant for e-LLE (Fig. 2b). Sequestration of the extractant to the organic phase was accomplished by functionalization of Fc with a hydrophobic saturated hydrocarbon tail (linked at the cyclopentadienyl ring of the Fc). Once oxidized to Fc+, the Fc site becomes highly hydrophilic and soluble in water (Fig. 2c). Therefore, to combat this phenomenon, the effect of functionalization with an increasing chain length on Fc was investigated with the aim of rationally designing a highly hydrophobic Fc with less than 1% loss to the aqueous phase when oxidized. The loss of oxidized Fc from the organic diluent DCM phase to the aqueous phase was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis for the following commercially available Fc functionalizations: Fc, 1,1′-diethylferrocene (diEtFc) and octylferrocene (OctFc). ddFc was synthesized in house to provide a highly hydrophobic Fc and to gauge the economics of redox extractant production. Without functionalization, 42% of the Fc was lost to the aqueous phase when oxidized (Fig. 2c). As the hydrocarbon side chain was lengthened, the Fc unit was more effectively sequestered to the organic phase, with only 0.6% of ddFc lost after vigorous sonication of the liquid phases. ddFc was thus chosen as the organic extractant for all subsequent experiments.

a, Comparison of control experiments for the recovery of gold, silver and copper, where the reversible metal recovery is the percentage of metal released divided by the initial metal mass. All control experiments used DCM as the organic-phase solvent. Metal recovery was only observed when ddFc and NOBF4 oxidizer were present. b, Rendering of oxidized ddFc+ binding to the dicyanoaurate anion. c, Percentage of Fc lost to the aqueous phase after oxidation as a function of the length of the hydrophobic tail. ddFc showed the least degree of loss. d, Recovery efficiency of gold for various aqueous reducing agents. For all experiments, 2 mM ddFc in DCM was oxidized using 2 mM NOBF4 and mixed with 5 mM KAu(CN)2. e, Cyclic voltammograms (CVs) at a scan rate of 10 mV s−1, with carbon felt working and counter-electrodes. The solution contained 10 mM aqueous sodium ferrocyanide for the green CV, and 10 mM aqueous sodium iodide for the red CV. The black CV was taken in a solution of DCM with 10 mM ddFc and 100 mM tetrabutylammonium hexafluorophosphate, and a leakless Ag/AgCl reference electrode was used. f, Gold uptake and release performance for various oxidizing agents. Data are presented as mean values ± s.d. with a sample size of 10.
A range of water-immiscible solvents (dibromomethane, chloroform, hexane, cyclohexane, xylene, 1-octanol, ethyl ether, butyl acetate, silicone oil and kerosene) were investigated as the organic extractant phase diluent for this study. As shown in Supplementary Fig. 16, halogenated solvents consistently achieved the highest recovery efficiency (>95%) of gold compared to the other investigated solvents, probably because of the high density differential with water, inertness over a wide potential window, and high relative permittivity while remaining immiscible with water. Hydrocarbon-based solvents (hexane, xylene, octanol and kerosene) showed the lowest gold uptake performance, averaging 151 ± 5 mg g−1, despite the low miscibility of these organic solvents in water (Supplementary Fig. 38) due to their low relative permittivities (less than 3; Supplementary Fig. 18). For the industrial use of e-LLE, it is necessary to recover any solvent entrained in the aqueous waste stream for economic and environmental reasons, particularly with halogenated solvents. This recovery is possible with a range of remediation methods43,44,45. In this study, DCM was chosen as the organic extractant phase diluent for its consistently high gold uptake of 300 mg g−1 and its recovery efficiency of >95% (Supplementary Fig. 16). However, a potentially greener alternative46,47 to DCM as the diluent for e-LLE is n-butyl acetate, which achieves a comparable gold uptake of 301 mg g−1 and 84% recovery efficiency. Future studies are envisioned where more environmentally benign solvents can be further explored and optimized for various metal-recovery applications.
Redox-mediator selection for the oxidizing loop
To enable the extraction of gold, the organic ddFc extractant was electrochemically oxidized from Fe(II) to Fe(III). The direct electrochemical redox of ddFc in organic DCM was possible with the addition of 100 mM tetrabutylammonium hexafluorophosphate supporting electrolyte (the ddFc voltammogram is shown in Fig. 2e). However, quaternary ammonium salts are known to increase the miscibility of halogenated solvents with water, resulting in the chemical loss of solvents (and the loss of both our ddFc extractant and a costly supporting electrolyte). Our design rationale thus focused on overcoming the low electrical conductivity of the organic-phase ddFc extractant by indirectly switching the ddFc redox state via two aqueous-phase redox-mediators. Two-phase liquid–liquid electrochemical systems containing redox-mediators have been studied previously within the context of electrolyzers and the enhancement of mass transfer for electrosynthesis48,49.
Aqueous triiodide can readily transform to diatomic iodine when exposed to an organic solvent50, and, when applied to our e-LLE system, iodine oxidant was able to oxidize ddFc and achieve a high gold uptake of 237 mg g−1 (Fig. 2f) with a very low, economical driving overpotential of 0.05 V (E1/2 = 0.46 V versus Ag/AgCl; Fig. 2e). When the concentration of triiodide oxidant was varied from 0 mM to 5 mM (1.25 mol I3− per ddFc), an increase in gold uptake was observed with a maximum of 198 mg g−1 at 5 mM I3−, and the experimental data matched the theoretical equilibrium Nernstian response (R2 = 0.98) of two-electron iodine reduction and one-electron ddFc oxidation (Fig. 3a). The ddFc oxidation with iodine was thus rapid, achieving equilibrium within 10 s. In the model, it was assumed that all oxidized ddFc+ would take up an equimolar amount of anionic gold species, and this assumption was probably true, implying that the molar utilization represents the percentage of ddFc oxidation for batch gold extraction results.

a, The molar utilization of ddFc as the molar ratio of aqueous triiodide oxidizer was increased for the experimental data (red) compared to the Nernst model (gray line). b, Schematic of the e-LLE system. c, Gold recovery efficiency of gold extracted with ddFc oxidized with equimolar NOBF4 as the molar ratio of aqueous ferrocyanide reducing agent was increased for the experimental data (blue) compared to the Nernst model (gray). d, Single-metal ddFc extraction isotherms for 5 mM solutions of gold, silver, copper and platinum. Experimental single-metal data are indicated by diamonds, and the Langmuir isotherm model predictions are shown as dashed lines for each. e, Gold recovery efficiency as a factor of the up-concentration ratio, set by decreasing the ratio of aqueous reducing solution to leach solution. As the volume of the reducing solution decreased, the gold was concentrated. f, ddFc cyclability study where the ddFc in DCM solution was recycled through ten consecutive extraction and release cycles. g, UV–vis spectra of the organic extractant solution containing 2 mM ddFc in DCM before oxidation (orange), after oxidation (blue) and extraction (red), and after reduction and release to the aqueous phase (black). h, Uptake and release performance of ddFc extraction with H2PtCl6, Na3IrCl6, H2IrCl6 and Na3RhCl6. Data are presented as mean values ± s.d. with a sample size of 10.
Redox-mediator selection for the reducing loop
Electrochemical release of the target metal bound to oxidized ddFc+ in the organic phase was possible via reduction of ddFc+. To accomplish this, an immiscible aqueous stream containing a suitable reversible redox species to mediate the reduction of ddFc had to be selected (Fig. 3b). Common reducing agents (10 mM) were tested (Na2S2O5, NaS2O3, ascorbic acid and Na4Fe(CN)6) to recover gold extracted by 2 mM ddFc in DCM (oxidized with 2 mM NOBF4). For sodium thiosulfate and sodium metabisulfite, 10 mM NaOH was added to the aqueous reduction/release solution to ensure facile electron transfer51. The resulting gold-recovery efficiencies (defined as the mass of released target species over the mass of extracted target, as a percentage) for various reducing agents are shown in Fig. 2d. All the reducing agents resulted in a color change in the organic phase from the blue of ddFc+ back to the yellow of reduced ddFc. Sodium thiosulfate had the lowest recovery efficiency of 70.3%, followed by sodium metabisulfite (80%) and ascorbic acid (81%). Sodium ferrocyanide achieved nearly complete gold recovery at 97.7 ± 1%, and the UV–vis spectra of the ddFc-containing organic phase after desorption with ferrocyanide contained only one absorption peak at 420 nm, indicative of reduced ddFc (Fig. 3g). Gold was recovered from 2 mM ddFc+ in DCM (with equimolar NOBF4) using aqueous ferrocyanide in a range of concentrations (0–20 mM). The resulting experimental gold-recovery efficiency data are compared to a Nernstian equilibrium model in Fig. 3c.
The gold-recovery efficiency increased from 0% to 96% when the ferrocyanide:ddFc ratio was at unity, and approached 98% with excess ferrocyanide. The Nernst model captured the experimental results well (R2 = 0.99). Accordingly, ferrocyanide rapidly reduced ddFc+ to ddFc, causing anionic dicyanoaurate to release from the organic phase and enter the aqueous ferrocyanide solution. The half-cell potential ferri/ferrocyanide redox couple was 0.21 V versus Ag/AgCl (Fig. 2e). Due to its high recovery performance, moderate reduction potential and electrochemical reversibility, sodium ferrocyanide was used at the redox couple to reduce ddFc+ for all gold capture and release experiments described in this work.
e-LLE performance for gold recovery
A gold absorption isotherm was constructed using 2 ml of 2 mM ddFc in DCM oxidized with equimolar NOBF4, as well as 1 ml of an aqueous KAu(CN)2 solution with concentration ranging from 0.1 mM to 20 mM. Gold extraction was carried out after shaking both immiscible liquids in a sealed vial for 10 s, and the gold-laden organic phase was transferred to a new vial containing 1 ml of aqueous 10 mM Na4Fe(CN)6 to reduce the ddFc and relinquish the captured gold to the aqueous reduction solution. The same process was carried out for the extraction targets KAg(CN)2 and K2Cu(CN)3, and the resulting reversible uptake isotherms showed favorability to gold, followed by silver, with negligible uptake of copper (Fig. 3d), agreeing with the trends from our previous work with PVF carbon nanotube (PVF-CNT) electrodes8,16.
For gold, the Langmuir isotherm model showed a close fit to the experimental results (R2 = 0.99), with a maximum uptake capacity Qmax of 253 mgAu gddFc−1 and an equilibrium coefficient of Keq = 39.4 mM−1 (Fig. 3d). The gold uptake approached a maximum beyond a gold concentration of 0.2 mM, indicating that the dicyanoaurate–ddFc binding was highly favorable (Supplementary Fig. 22a). Below a concentration of 0.2 mM, over 99% of gold was removed within a single extraction pass, and above 0.2 mM, 81% of the ddFc was utilized for the uptake of gold, proving ddFc to be a high-performance gold extractant with high atom efficiency (Supplementary Fig. 22b). When the concentration of ddFc in the organic phase was varied from 1 mM to 20 mM with the concentration of aqueous KAu(CN)2 held constant (10 mM), the resulting gold absorption isotherm matched the Langmuir model (Supplementary Fig. 23), confirming that a Langmuir adsorption behavior captures the gold–ddFc extraction equilibrium closely.
Gold capture was highly reversible over a range of gold concentrations from 0.1 to 20 mM (Supplementary Fig. 24) and ddFc concentrations from 1 mM to 20 mM (Supplementary Fig. 25), with over 90% recovery efficiency. ddFc (2 mM) in DCM was reused over ten extraction and release cycles with 5 mM KAu(CN)2, and the gold uptake remained consistently high (uptake capacity of 304 mg g−1), with a slight loss of utilization from 86% to 72% by the tenth reuse cycle (Fig. 3f), which may be explained by losses during liquid handling in the batch process. The gold-recovery efficiency, however, remained consistently high (recovery efficiency of 94%), indicating that the ddFc was successfully regenerated each cycle and was capable of repeated turnover.
Up-concentration of gold with the ddFc e-LLE system was investigated by changing the volume ratio of the aqueous leach and aqueous release solutions from 1:1 to 16:1 for a theoretical gold up-concentration ratio of 16. After extracting 1 mM KAu(CN)2 with 2 mM oxidized ddFc+ in DCM and releasing the organic-bound gold into aqueous ferrocyanide solution, the gold-recovery efficiency was 97 ± 4% for all up-concentration ratios of gold (Supplementary Fig. 26). Extraction of 235 mg l−1 of gold from a 16-ml solution and subsequent desorption to a 1-ml solution yielded a final gold concentration of 3,012 ± 7 mg l−1 (Fig. 3e) with a recovery efficiency of 92%, demonstrating that up-concentration was possible with the e-LLE system.
Multicomponent selectivity in e-LLE
Adsorption isotherms were constructed for KAg(CN)2 and K2Cu(CN)3 over a range of concentrations from 0.1 mM to 20 mM with 2 mM ddFc in DCM oxidized with equimolar NOBF4. The reversible uptake isotherms of silver and copper are compared to those of gold in Fig. 3d. Silver uptake with ddFc was highly irreversible, with an average recovery efficiency of 35%, indicating that ddFc extraction was not the major mechanism of silver removal. In control experiments, silver extraction only occurred with oxidized ddFc+ (Fig. 2a). Therefore, oxidized ddFc+ was necessary for silver removal, but binding of the Ag(CN)2− to ddFc+ in DCM solvent may have resulted in decomposition of the silver complex to AgCN, which is insoluble in water52. The reversible uptake isotherm for silver did fit the Langmuir absorption isotherm model (R2 = 0.97), with a Qmax of 83 mg g−1 and Keq of 1.2 mM−1, indicating that ddFc–silver binding did occur. When compared to the dicyanoaurate absorption isotherm in Fig. 3d, uptake of anionic dicyanoargentate was indeed unfavorable with ddFc, despite the structural similarities of the cyano–silver and –gold complexes. In a binary mixture of 5 mM KAu(CN)2 and 5 mM KAg(CN)2, the selectivity factor of gold extraction with ddFc, relative to silver, was 18.5:1 (Supplementary Fig. 27), confirming the favorability of gold–ddFc binding over that of silver.
Copper uptake with ddFc was largely irreversible, with an average recovery efficiency of 15% and a maximum reversible uptake of 6.4 mgCu gddFc−1 at 10 mM K2Cu(CN)3 (Supplementary Fig. 28), so copper binding to ddFc+ was highly unfavorable. In control experiments, copper extraction only occurred with oxidized ddFc+ (Fig. 2a), indicating that copper indeed interacted with ddFc+, albeit to a small degree (1% extraction). Binary extraction of 5 mM gold and 5 mM copper resulted in a high gold selectivity factor of 113:1 relative to copper, as expected (Supplementary Fig. 29). For stability of the cyano–copper complex, 5 mM KCN was included in the aqueous leach solution, and so the effect of excess free cyanide (KCN) in the metal leach was investigated with 5 mM KAu(CN)2 as the target metal. The uptake of gold was observed to decrease progressively from 301 to 165 mgAu gddFc−1 as the concentration of KCN increased from 0 to 10 mM, but the gold-recovery efficiency remained consistently high with an average of 90% (Supplementary Fig. 30). From the literature, it can be seen that free cyanide (CN−) can be oxidized to (CN)2 above a formal potential of 0.35 V versus Ag/AgCl53. Therefore, excess free cyanide probably caused premature reduction of ddFc+, with a formal potential of 0.41 V versus Ag/AgCl, resulting in lower gold uptake. However, a great excess (>10 mM) of free cyanide was required to appreciably reduce ddFc+, and 5 mM free KCN resulted in only a 13% loss in gold uptake. As such, it is recommended that the free cyanide concentration of a metal leach solution should be less than 5 mM.
PGM recovery performance
Extractions of various anionic chloro-complexes of PGMs were tested with ddFc e-LLE ([Pt(IV)Cl6]2−, [Ir(III)Cl6]3−, [Ir(IV)Cl6]2− and [Rh(III)Cl6]3−). For each species, 5 mM aqueous PGM was extracted into DCM by 2 mM ddFc oxidized with equimolar NOBF4, and subsequently released to a new aqueous reduction stream containing 10 mM sodium ferrocyanide for complete reversible extraction. Pt(IV) and Ir(IV) showed the highest uptakes (124 mgPt gddFc−1 and 144 mgIr gddFc−1) and the highest separation reversibilities of 81% and 86%, respectively (Fig. 3h). The uptake of Ir(III) was only 71 mgIr gddFc−1—half that of Ir(IV)—and virtually no Rh(III) was extracted by ddFc (Fig. 3h). These findings suggest that ddFc-enabled extraction most favorably extracts anionic metal complexes of increasing atomic size, that is, Au > Pt ~ Ir > Ag > Ru > Cu, and uptake decreases as the formal charge of the target anion becomes more negative, that is, [Au(CN)2]− > [PtCl6]2− > [IrCl6]3−. Previous density functional theory (DFT) studies have shown that selective Fc–metal complex binding is largely based on charge-transfer interactions8,16, with transition-metal complexes displaying preferential binding with Fc over competing inorganic anions, supporting the selectivity trends observed with ddFc. A reversible uptake isotherm was constructed for chloroplatinic acid, and the Langmuir isotherm model was fit (R2 = 0.988; Qmax = 148 mg g−1 and Keq = 0.88 mM−1) (Fig. 3d and Supplementary Fig. 31). Notably, the Qmax of [PtCl6]2− was roughly half that of [Au(CN)2]−, probably due to the Pt complex’s formal charge of 2−, requiring two ddFc+ units to extract a single Pt species.
Aqueous PGMs were mixed with pure solvents (DCM, chloroform, hexane and xylene) to benchmark the background uptake due to the PGM–organic solubility, and it was found that only 4.1% of the aqueous PGM salts leached to the organic DCM phase on average (Supplementary Fig. 32). The Pt(IV) and Ir(IV) complexes were more soluble in all organic solvents compared to Ir(III) and Rh(III), and, similar to the uptake results, this trend was probably due to the lower 2− formal charge of [PtCl6]2− and [IrCl6]2−. The uptake of 5 mM aqueous PGM salts with reduced ddFc (no oxidizer) was carried out, and, despite the lack of NOBF4 oxidizer, 75 mg g−1 of Ir(IV) was reversibly extracted, whereas all other PGM salts showed negligible uptake. The reversible uptake of each PGM species with and without NOBF4 oxidizer is shown in Supplementary Fig. 33. Iridium had a reversible redox couple, IrCl62−/IrCl63−, with a formal potential of 0.65 V versus Ag/AgCl, which allowed Ir(IV) to spontaneously oxidize ddFc to ddFc+ (E0 = 0.41 V versus Ag/AgCl) and simultaneously bind the now reduced Ir(III) to oxidized ddFc+. Therefore, it was possible to selectively and spontaneously capture H2IrCl6 using ddFc e-LLE (requiring no energy or material consumption).
Continuous redox-mediated ion-selective extraction
A 20-ml-min−1 stream of 5 mM KAu(CN)2 was continuously extracted and purified without chemical consumption using our e-LLE system (Supplementary Figs. 13−15). The system consisted of three closed liquid loops, three simple solvent extraction columns, and an electrochemical flow cell with two flow paths separated by a cation exchange membrane (CEM). A 20-ml volume of 2 mM ddFc in dibromomethane (DBM) organic solvent was continuously cycled in a closed loop at 10 ml min−1 through three solvent extraction columns: oxidizing, leach and reducing columns (Fig. 4a). In the oxidizing column, ddFc was oxidized to the active binding extractant species, ddFc+, by a closed aqueous oxidizing loop of 10 mM I3− flowing at 20 ml min−1, where the reduced iodide was electrochemically regenerated. After oxidation to ddFc+, the organic extractant phase entered the leach column, alongside the 20 ml min−1 aqueous gold leach stream, initially containing 10 mM KAu(CN)2. The 20-ml gold leach stream was continuously recirculated, and 92% of aqueous gold was extracted (Supplementary Fig. 34) with an impressive gold uptake of 1,402 mg gddFc−1, achieved over a period of 8 h (Fig. 4b).

a, Schematic of the continuous-flow e-LLE system used for continuous-flow extraction experiments. b, Continuous e-LLE uptake and gold recovery performance kinetics. c, Energy consumption normalized by the mass of gold that was recovered, and cumulative charge over time for the flow cell as it regenerated oxidizer and reductant. Data are presented as mean values ± s.d. with a sample size of 10.
Following the leach column, the gold-laden organic phase passed through the final reducing column, where a 10-ml closed aqueous loop of 10 mM sodium ferrocyanide (20 ml min−1) simultaneously reduced the ddFc+ and extracted the now unbound [Au(CN)2]− anion into the aqueous reduction loop. The organic ddFc was then cycled back to the oxidation column to repeat the e-LLE process, thereby forming a closed extractant loop that did not consume extractant or solvent. All extracted gold was ultimately sequestered to the closed aqueous reduction loop, where it was concentrated. A total of 92% of the extracted gold was reversibly recovered (Fig. 4b) and up-concentrated by a factor of 2, and 3.7 gold molecules were extracted per ddFc molecule, demonstrating that the ddFc extractant was indeed recycled and reused (Supplementary Fig. 34).
Finally, the spent iodine oxidizing agent and ferrocyanide reducing agent were electrochemically regenerated in a CEM-separated flow cell with 4 × 4 × 0.3-cm carbon felt electrodes (a configuration similar to that of a redox flow battery)54,55,56. The cell was operated with a constant current of 2 mA until a two-electrode potential of 0.23 V (Eox − Ered) was reached, then the potential was held constant thereafter (Supplementary Fig. 35). Due to the low operating potential of <0.23 V and operating current of <2 mA, coupled with the high molar utilization of ddFc (3.7 mol Au/ddFc), the energy consumption per gold recovered was 0.143 kJ gAu−1 (Fig. 4c), lower than that of our previous PVF-CNT work19 by a factor of 75 (98.7% reduction in energy consumption). In terms of cost, our continuous e-LLE system had a total energy cost of US$2.89 per metric ton of gold recovered, with no materials consumed. For perspective, the solar cell from a pocket calculator (5 cm2, 3-mW power) could recover 2.2 g of pure gold per day with our unoptimized bench-scale e-LLE proof-of-concept system.
Evaluation of e-LLE for real metal leach streams
A 310-g sample of locally sourced electronic waste in the form of DDR3 computer RAM modules was added to 1 l of 10 mM KCN to leach the surface gold and other metals, following the optimized leaching procedure of our previous work19. The leach process was optimized to minimize chemical consumption. After 24 h of aerated leaching, the e-waste leach solution contained 2.11 g l−1 copper, 676.53 mg l−1 nickel, 309.89 mg l−1 gold, 1.94 mg l−1 iron and 0.13 mg l−1 silver as the five major constituents (Supplementary Fig. 36a), and the leach solution had a pH of 10. Without any further processing of the leach solution, the gold was recovered using our e-LLE process with 2 mM ddFc electro-extractant, achieving a gold uptake of 101 mg g−1 (Supplementary Fig. 36a). When compared to the pure gold absorption isotherm in Fig. 3d, gold uptake from the e-waste leach was 30% lower; this loss in uptake was probably due to the 29-fold molar excess of competing copper and nickel ions in the leach solution. When the organic phase ddFc+ was reduced with 10 mM of aqueous ferrocyanide, 91.6% of extracted gold was released into the aqueous reducing phase, and the gold was successfully purified from 9.9% to a final gold purity of 59.6% in a single extraction pass (Fig. 5a), with a gold selectivity factor of 13.3:1 relative to copper, nickel, iron and silver, proving that selective recovery and purification of gold was possible with real-world e-waste leach solutions with the ddFc electro-extractant e-LLE system.

a, Comparison of the initial metal purity (white circles) of a gold leach solution from electronic waste and the final purity (blue circles) obtained after e-LLE extraction. b, McCabe–Thiele plot for e-LLE, where the upper equilibrium line was constructed using the gold selectivity factor relative to silver, copper, nickel and iron. The electronic waste leach solution containing 10% gold was purified with three consecutive e-LLE stages. Xi refers to the initial gold purity (wt%), and Xf refers to the final metal purity (wt%). c, Comparison of the initial metal purity (white circles) of a gold leach solution from simulated mining ore and the final purity (blue circles) obtained after e-LLE extraction. d, Photograph of a 2014 Scion Tc catalytic converter and the monolithic catalyst structure that PGMs were extracted from to produce a real-world PGM leach solution. e, e-LLE extraction performance of PGMs from the catalyst converter leach solution. The gray bars represent the change in metal purity following e-LLE, and the blue diamonds represent the selectivity factor of ddFc.
Gold in the e-waste leach solution was further purified by consecutive extractions using the e-LLE approach; the first extraction stage increased the gold purity from 10% to 60.1%, the second extraction increased it to 91.4%, and a third extraction brought the final gold purity to 99.8% (Fig. 5b). A McCabe–Thiele plot was constructed from the multi-stage e-LLE experimental results, and the e-LLE equilibrium line was constructed from the system mass balance and the average gold selectivity factor of 20 (relative to silver, copper, iron and nickel competing species). Figure 5b shows that the experimental multi-stage performance is captured within 5% error using the McCabe–Thiele graphical approximation method for the theoretical number of separation stages. Therefore, Fig. 5b may be used as a process design roadmap to predict the number of consecutive e-LLE stages required to achieve a particular gold purity, and, using this method, bullion-grade gold (99.99% pure) could be achieved from 10% gold e-waste leach in a total of four e-LLE stages. Furthermore, a multi-stage countercurrent cascade of equilibrium stages can be feasibly realized with conventional multi-tray solvent extraction architectures57.
A simulated gold-ore leach solution was prepared following methods described previously in the literature19, resulting in a solution containing 21.17 mg l−1 Cu, 19.31 mg l−1 Fe, 1.73 mg l−1 Au, 1.37 mg l−1 Ag and 0.78 mg l−1 Ni. After e-LLE extraction with 2 mM ddFc, 98.3% of the gold contained in the leach solution was extracted (Supplementary Fig. 36b) with a relative selectivity factor of 50.1, and the purity of the gold increased from 3.9% in the leach to a final gold purity of 67.0% (Fig. 5c), demonstrating the applicability of our e-LLE approach for gold-ore refinement.
Finally, a real PGM leach solution was generated from the digestion of an automotive catalytic converter (2014 Scion Tc (Fig. 5d)), and PGM purification was carried out with e-LLE. Material from a new 2014 Scion Tc catalyst was ground and leached with chlorine gas as the oxidizer and Cl− as the stabilizing ligand, resulting in an acidic aqueous solution containing trace anionic chloro–PGM complexes (0.92 mg l−1 Ir, 0.46 mg l−1 Pt and 0.03 mg l−1 Rh), with the major species being palladium at 1,471.6 mg l−1. Despite the high initial palladium concentration, iridium and platinum were the only two PGM species to increase in purity after e-LLE (Fig. 5e), with platinum increasing from 0.1% to 9.7% pure in a single extraction stage, and with a platinum selectivity factor of 294 relative to Pd + Ir + Rh. Therefore, the e-LLE system demonstrated selective purification of platinum from a real-world PGM leach stream.
Techno-economic analysis
The techno-economics of our e-LLE system were evaluated and compared to standard industrial technology, activated carbon (AC)-based carbon in pulp (CIP)32, and our previous work utilizing a PVF-CNT electrode adsorbent. The scope of this techno-economic analysis covered all processing of the gold stream following the cyanide leaching stage, including the recovery and concentration of gold in solution and gold electrodeposition (electrowinning; Fig. 6b). From the basis of a leach solution flow of 4,000 l min−1 containing 50% gold and silver at a concentration ranging from 0.006 to 1,000 mg l−1 gold, the energy and materials costs were estimated for each recovery method—CIP, PVF electrosorption and e-LLE, utilizing a six-capture-unit cascade model, which recovered 99.6% of gold from the leach solution19. For the e-LLE system, the cost to make up the 0.1% of solvent as well as the 0.5% of ddFc lost to the aqueous tailings stream were the dominant cost factors at an initial gold leach concentration below 10 mg l−1 (low-grade ore-mining conditions), justifying the selection of a solvent and extractant with high immiscibility in water (Supplementary Fig. 38). At a gold feed concentration above 10 mg l−1, the energy cost of gold electrodeposition was dominant, and the energy cost of continuously extracting gold with ddFc was lowest, making up 10% of the total operational cost of ddFc due to the low 0.86 kJ gAu−1 energy consumption of the e-LLE process (Fig. 6a). At a gold feed concentration of 100 mg l−1, our ddFc system cost US$0.49 per kg of gold recovered, compared to US$5.90 per kg for PVF electrosorption and US$245 per kg for conventional AC (Fig. 6a). Therefore, the e-LLE system can be >500 times lower in cost than the current industrial process, CIP, as a result of consuming over 99.9% less energy and 99.4% less chemical materials. Conventional AC was estimated to consume 14,000 l of fresh water per kilogram of gold recovered, accounting for 35% of the total utility cost (US$87 per kgAu). In addition, the high selectivity of the e-LLE process resulted in 99.2% pure gold, whereas CIP resulted in 50% gold, requiring further costly processing that is not covered in this analysis. Finally, the e-LLE process is a fully continuous process, unlike CIP, which would simplify the overall process design and reduce the process footprint, personnel hours and system downtime27, which could increase sustainability and reduce the overall costs of gold recovery.

a, A cost comparison and breakdown for the three methods of gold recovery: AC (the industrial standard used in the CIP process), PVF-CNT electrosorption based on earlier work by Cotty and colleagues19, and the proposed ddFc e-LLE extraction. The y axis is logarithmic. b, Schematic of all inputs and outputs within the bounds of this techno-economic analysis (TEA).
Discussion
We have demonstrated a fully continuous approach to selective electrochemical separation, utilizing a ddFc redox-active extractant in an e-LLE. The electroactive ddFc extractant was found to selectively extract gold, platinum and iridium with highly enhanced molar utilization of the ferrocenium (>89%), while retaining >99% gold-recovery efficiency owing to the continuous nature of the e-LLE system. By using reducing and oxidizing redox-mediators with a low 0.23-V working potential, the energy consumption of e-LLE was over three orders of magnitude lower than that of conventional CIP (EddFc = 0.86 kJ gAu−1, ECIP = 2,046 kJ gAu−1). ddFc was observed to be highly selective to gold (>20:1) and capable of simultaneously providing gold purification to over 99% and up-concentration by a factor of 16. ddFc was observed to preferentially bind to metal anions of increasing atomic mass, that is, Au > Pt ~ Ir > Ag > Ru > Cu, and uptake decreased as the formal charge of the target anion became more negative, that is, [Au(CN)2]− > [PtCl6]2− > [IrCl6]3−.
Real-world precious-metal leach streams were produced from electronic waste, simulated gold-mining ore and automotive catalytic converter material, and the e-LLE process was found to be highly efficient for purifying gold, platinum and iridium directly. In addition, the e-LLE system showed great potential to operate with greener solvents such as n-butyl acetate, while retaining its highly selective and continuous metal purification capabilities. Finally, the e-LLE system was operated continuously, simultaneously extracting and releasing gold, to achieve over 1,500 mg g−1 uptake, over 90% gold removal, and 91% gold-recovery efficiency. A techno-economic analysis further indicated that the ddFc e-LLE process was highly favorable from a process perspective (US$0.49 per kgAu). We envision that the electrochemically mediated LLE is a step change and demonstrates the versatility and applicability of electrochemical approaches for broader, continuous molecularly selective separations.
Methods
Materials and instrumentation
All chemicals and solvents used in this work were obtained from Sigma Aldrich, VWR, Fisher Scientific or TCI.
Flash chromatography was performed using a Büchi Pure C-810 chromatography system with Büchi Pureflex Ecoflex silica cartridges as the stationary phase. A CHN analyzer (CE440, Exeter Analytical) was used to measure the composition of carbon, hydrogen and nitrogen in compounds. Samples were burned at a high temperature (1,800 °C) with a He purge. The products mixed with reagents were analyzed by a series of thermal conductivity detectors for precision measurement of C, H and N elements in organic compounds58. NMR spectra were recorded on Varian Unity Inova 400-MHz or 500-MHz spectrometers, both equipped with a Nalorac QUAD probe. Electrospray-ionization mass spectrometry (ESI-MS) was performed with a Waters Q-TOF Ultima ESI.
Synthesis of ddFc
The synthesis of ddFc was carried out in a 1 l three-neck flask with a condenser and dripping funnel under an Ar atmosphere. First, 28.38 g (212.9 mmol or 2.2 equiv.) aluminum trichloride was suspended in 200 ml of anhydrous DCM. Separately, 42.33 g (45.9 ml, 193.5 mM or 2 equiv.) dodecanoyl chloride was added to 100 ml of DCM and slowly added. The mixture was cooled in an ice bath, and a solution of 18 g (96.76 mmol or 1 equiv.) Fc in 120 ml of DCM was added. The mixture was stirred for 16 h at room temperature. The reaction mixture was again cooled in an ice bath and a solution of 7.321 g (193.5 mmol or 2 equiv.) sodium borohydride in 100 ml of triethylene glycol dimethyl ether was added slowly. The mixture was stirred for 4 h.
The reaction was quenched by adding 500 ml of 1 M HCl, and the phases were separated. The aqueous phase was extracted with DCM and combined with the organic phase, and the product containing organic phase was washed with water, saturated NaCl solution and dried with MgSO4. The solvent was evaporated to yield a crude ddFc product. The product was purified by flash chromatography with hexane. The final yield of ddFc was 25.17 g (50%).
1H NMR (500 MHz, CDCl3) δ 4.08–3.89 (m, 8H), 2.32 (t, J = 6.6 Hz, 4H), 1.51 (t, J = 5.5 Hz, 4H), 1.41–1.20 (m, 38H), 0.91 (t, J = 6.7 Hz, 6H).
13C NMR (126 MHz, CDCl3) δ 89.42, 68.57, 67.58, 31.84, 31.19, 29.61, 29.56, 29.47, 29.31, 29.27, 22.60, 14.02.
HR-ESI (m/z): [M]+ calculated for C34H58Fe, 522.3888; found: 522.3875. Elemental analysis (%): calculated for C34H58Fe: C 78.13, H 11.18; found: C 78.08, H 11.14.
Determination of total metal concentration
The total metal concentrations of Au, Ag, Cu, Fe, Ni, Pt, Pd, Ir and Rh in the aqueous phase were determined with an Agilent 5100 series ICP-OES system. Samples were diluted 50:1 with deionized (DI) water (metal cyanide solutions) or 5% HCl solution (chloro–metal solutions). For each element, a four-point calibration curve was created using certified metal standard solutions, and linearly fit with R2 > 0.999. No interference among element species was encountered. For batch-scale e-LLE experiments, a 100-μl sample was taken before and after extraction and release, for a total of four samples per single recovery experiment. For continuous-flow operation, a sample was taken from each aqueous stream (oxidizing, leach and reducing streams), and samples were taken over time.
UV–vis experimentation
UV–vis absorption spectra were measured with an Agilent Cary 60 UV–vis double-beam spectrophotometer (Agilent Technologies) in a range of 200–800 nm. Samples were transferred (using a 1-ml disposable plastic pipette) into a cuvette, which was placed into the spectrophotometer. At the beginning of each experiment, the absorbance of a blank sample containing only the solvent was measured. The absorbance for the cuvette containing the sample was also measured, and the wavelength at the absorption peak was designated as the wavelength of the sample and compared with literature.
A 10-mm Hellma quartz glass cuvette with DCM as a solvent was used for ddFc. A calibration curve was constructed for ddFc by recording the spectra of known concentrations of ddFc in DCM (blank, 1, 2, 3, 4 and 5 mM). The resulting absorbance at a characteristic wavelength of 438 nm was plotted against the analyte concentration, resulting in a linear fit with an R2 value of 0.999 (Supplementary Fig. 5). UV–vis spectra were also used to characterize the ddFc via the extraction and release cycle: 2 mM ddFc in DCM was measured as is, after oxidation with 2 mM NOBF4, after extraction with oxidized ddFc, and after reduction and desorption.
A 1-mm Hellma TrayCell quartz glass cuvette with Factor 10 and water as a solvent was used for the triiodide. The absorbance spectra gave two distinct peaks with maxima at 290 nm and 352 nm, correlating with data for the triiodide anion from the literature (Supplementary Fig. 6). A linear calibration curve was also made, with R2 values of 0.9922 and 0.9918 for the peaks, respectively (Supplementary Figs. 7 and 8).
Batch-scale e-LLE experimentation
For batch-scale extraction experiments, two vials were used (labeled vial A and vial D, respectively). A 2 ml volume of 2 mM ddFc in DCM was added to vial A along with 40 μl of 100 mM NOBF4 in acetonitrile. After 30 s (to allow the ddFc to become fully oxidized, as visually determined by deep blue coloring), 1 ml of aqueous leach solution was added to vial A. The most common example of an aqueous leach solution is 5 mM KAu(CN)2 in DI water. The two immiscible liquids in vial A were vortex-mixed at 3,000 r.p.m. for 10 s, and typically given 1 min to phase-separate. Samples of the aqueous phase before and after mixing were taken to determine the amount of metal extracted to the organic phase using ICP-OES.
Vial D was filled with 1 ml of 10 mM K4Fe(CN)6 aqueous reducing agent, and 1.5 ml of the organic phase of vial A was pipetted into vial D. The two immiscible liquids in vial D were vortex-mixed at 3,000 r.p.m. for 10 s, and typically given 1 min to phase-separate. The blue organic phase rapidly turned back to yellow within the first second of mixing, visually confirming that reduction of ddFc had occurred. Samples of the aqueous phase before and after mixing were taken to determine the amount of metal released from the organic phase using ICP-OES.
Continuous-flow cell and extraction column design
Triple column apparatus design
The extraction column apparatus (the triple column) was designed to be three-dimensionally (3D) printed with both fused deposition modeling (FDM) and stereolithography (SLA) printers. All three columns (oxidizing, leach and reducing) were combined in a single unit to minimize process control of the three liquid–air and three liquid–liquid interfaces. The interface control is shown in Supplementary Fig. 10, where the liquid–air interfaces are seen to be passively controlled via weirs to an overflow reservoir. The triple column was designed to link the liquid–liquid interface between the aqueous and organic phases of all three columns to the liquid level of the organic-phase reservoir. Accordingly, the three liquid–liquid interfaces are easily controlled by maintaining a single interface. All cells of the triple column were exposed to the atmosphere, allowing the liquid pressures to equilibrate. The triple column used for the experimental runs was 3D-printed with Fluorinar-C PVDF on a Prusa Research i3 MK3s system in a temperature-controlled enclosure.
To combat the slow evaporation of organic solvent, whose liquid level controls the liquid–liquid interface height for all three columns, an auxiliary solvent reservoir was used with an electronic level-control sensor. If the level of the auxiliary solvent reservoir dropped below the setpoint, more pure solvent was automatically pumped to raise the liquid level, thereby maintaining the critical liquid–liquid interface of each solvent extraction column (Supplementary Fig. 11). Automation was accomplished using an RP2040 microcontroller board, a stepper motor with peristaltic pump head and a TMC 2209 stepper motor driver (shown in Supplementary Fig. 14).
Continuous-flow-through electrochemical cell design
The electrochemical cell used to simultaneously replenish the oxidizing and reducing stream was inspired by a redox flow battery cell design (Supplementary Fig. 13). The cell consisted of titanium current collectors and 40 mm × 40 mm AvCarb G475 carbon felt electrodes with a working thickness of 3 mm (35% compression). A Nafion 115 CEM was used to isolate the two channels of the flow cell. The liquid flow path was designed to enter through a hole in the corner of the current collector, and, within the flow path spacer, the liquid was forced to flow through the length of the compressed carbon felt electrode to the other side, where the liquid exited from a second hole in the current collector. The backing plates were constructed with an Elegoo Mars 3 Pro SLA 3D printer using clear photo-resin. The flow path spacers were 3D-printed with a Prusa Research i3 Mk3s FDM 3D printer using Fluorinar-C PVDF filament. All gaskets were Viton rubber.
Continuous-flow operational procedure
The in-house designed and 3D-printed continuous-flow apparatus containing three solvent extraction columns, three aqueous reservoirs and one organic extractant reservoir was connected to four peristaltic pumps and the in-house designed flow cell, with silicone tubing for all aqueous lines and Viton tubing for the organic extractant loop, as shown in Supplementary Fig. 11.
The system start-up procedure was as follows. First, the organic reservoir of the triple column was filled with 20 ml of pure DBM organic solvent, the aqueous oxidizer reservoir with 15 ml of 100 mM NaI, the aqueous leach reservoir with 15 ml of 10 mM KAu(CN)2 and the aqueous reducing reservoir with 15 ml of 100 mM Na3Fe(CN)6. Pumps 1, 2, 3 and 4 were turned on to purge air from the lines and begin equilibrating the liquid–liquid interfaces in each extraction column. The flow rates of pumps 1, 2 and 3 were 10 ml min−1, and pump 4 had a flow rate of 5 ml min−1. Additional pure organic solvent was added to the organic extractant reservoir until the liquid level was sensed by the level controller, signifying that all liquid–liquid interfaces were correctly set (the typical total solvent volume was 37 ml). Once the liquid interfaces were within range (halfway between full and empty), the interface control loop was activated. The interface control loop combatted the unavoidable slow evaporation of organic solvent by adding additional pure solvent with pump 5, only when the liquid level fell below the setpoint, using a microcontroller. Finally, 0.75 ml of 100 mM ddFc extractant in DBM was added to the organic reservoir and allowed to mix, resulting in a total ddFc concentration of 2 mM in the organic extractant loop. After 5 min of equilibration, start-up was completed.
For the experimental runs, the total gold concentration in the oxidizing, leach and reducing loops was monitored by ICP-OES by taking periodic 50 μl samples from the respective reservoirs. The 50 μl samples were diluted in 5 ml of DI water to achieve a 1:100 dilution. An experiment began by first monitoring the open-circuit potential of the membrane flow cell for 5 min. The continuous capture and release of target species with e-LLE was initiated by applying a constant 2 mA current until the overall cell potential reached 0.23 V, which was then held constant at that potential. The system was operated in an ‘overdamped’ configuration to allow the oxidizing and reducing loop to achieve steady-state concentrations without spikes. For system shutdown, the membrane cell was set to an open-circuit potential for an hour to allow the system to achieve equilibrium once again. Throughout the entire experimental run, samples were periodically taken from the aqueous reservoirs to monitor the total gold concentration with ICP-OES (the volume change from sampling was noted).
Electronic waste leaching procedure
We used 200-g samples of as-received DDR3 RAM modules from the University of Illinois Surplus Equipment Department, which were added to a 1-l aqueous solution containing 10 mM KCN and 1 mM KOH. Air was bubbled at 3.5 l min−1 into the RAM-containing leach vessel for 24 h. The air passed through a series of bubble traps containing 1 M KOH: first before the leaching vessel to humidify the air and remove acidic CO2, and second after the leach vessel to mitigate the release of hydrogen cyanide. The cyanide leaching apparatus was housed in a fume hood with secondary containment. After 24 h of air bubbling, there were no visible signs of gold plating remaining on the RAM modules, and the gold-containing leach solution was decanted off to a sealed container. No further processing or filtration was done to the leach solution, which was used for recovery experiments as is.
PGM leaching procedure from an automotive catalytic converter
A catalytic converter from a 2014 Scion Tc was purchased new from Toyota, and the PGM-containing catalyst material was removed from the stainless-steel housing. A total of 600 g of catalytic converter material was recovered, and blended to a powder to achieve homogeneity. For leaching, 900 ml of 38% HCl was added to 100 g of catalytic converter material in a 1 l glass bottle. Chlorine gas was generated in situ by slowly adding 100 ml of 9% sodium hypochlorite solution (household bleach). The bottle was capped and vigorously stirred, and after 24 h, the remaining solids were filtered out, leaving a clear red solution. Any remaining chlorine gas was purged by bubbling air through the leach solution. After another 24 h, the solution was topped up to 1 l with DI water (~50 ml was added) and used as is for PGM recovery experiments.
Data availability
All data are available within the paper and the Supplementary Information. Source data are provided with this paper.
References
Srimuk, P., Su, X., Yoon, J., Aurbach, D. & Presser, V. Charge-transfer materials for electrochemical water desalination, ion separation and the recovery of elements. Nat. Rev. Mater. 5, 517–538 (2020).
Alkhadra, M. A. et al. Electrochemical methods for water purification, ion separations and energy conversion. Chem. Rev. 122, 13547–13635 (2022).
Su, X. et al. Asymmetric Faradaic systems for selective electrochemical separations. Energy Environ. Sci. 10, 1272–1283 (2017).
Kim, K., Raymond, D., Candeago, R. & Su, X. Selective cobalt and nickel electrodeposition for lithium-ion battery recycling through integrated electrolyte and interface control. Nat. Commun. 12, 6554 (2021).
High, M. et al. Precursor engineering of hydrotalcite-derived redox sorbents for reversible and stable thermochemical oxygen storage. Nat. Commun. 13, 5109 (2022).
Li, X., Zhao, X. H., Liu, Y. Y., Hatton, T. A. & Liu, Y. Y. Redox-tunable Lewis bases for electrochemical carbon dioxide capture. Nat. Energy 7, 1065–1075 (2022).
Liu, Y. Y., Ye, H. Z., Diederichsen, K. M., Van Voorhis, T. & Hatton, T. A. Electrochemically mediated carbon dioxide separation with quinone chemistry in salt-concentrated aqueous media. Nat. Commun. 11, 2278 (2020).
Cotty, S. et al. Electrochemical recycling of homogeneous catalysts. Sci. Adv. 8, eade3094 (2022).
Massen-Hane, M., Diederichsen, K. M. & Hatton, T. A. Engineering redox-active electrochemically mediated carbon dioxide capture systems. Nat. Chem. Eng. 1, 35–44 (2024).
Wilcox, J. An electro-swing approach. Nat. Energy 5, 121–122 (2020).
Kim, N., Elbert, J., Kim, C. & Su, X. Redox-copolymers for nanofiltration-enabled electrodialysis. ACS Energy Lett. 8, 2097–2105 (2023).
Kim, N., Lee, J. & Su, X. Precision tuning of highly selective polyelectrolyte membranes for redox-mediated electrochemical separation of organic acids. Adv. Funct. Mater. 33, 2211645 (2023).
Su, X., Chen, Z., St-Pierre, J. & Vasiljevic, N. Electrochemistry for recycling. Electrochem. Soc. Interface 30, 41–43 (2021).
Chen, R., Sheehan, T., Ng, J. L., Brucks, M. & Su, X. Capacitive deionization and electrosorption for heavy metal removal. Environ. Sci. Water Res. Technol. 6, 258–282 (2020).
Su, X. & Hatton, T. A. Redox-electrodes for selective electrochemical separations. Adv. Colloid Interface Sci. 244, 6–20 (2017).
Chen, R. L. et al. Structure and potential-dependent selectivity in redox-metallopolymers: electrochemically mediated multicomponent metal separations. Adv. Funct. Mater. 31, 2009307 (2021).
Kim, K. et al. Electrochemical approaches for selective recovery of critical elements in hydrometallurgical processes of complex feedstocks. iScience 24, 102374 (2021).
Candeago, R. et al. Semiconducting polymer interfaces for electrochemically assisted mercury remediation. ACS Appl. Mater. Interfaces 12, 49713–49722 (2020).
Cotty, S. R., Kim, N. & Su, X. Electrochemically mediated recovery and purification of gold for sustainable mining and electronic waste recycling. ACS Sustain. Chem. Eng. 11, 3975–3986 (2023).
Guo, Z.-Y. et al. Development of electrochemical lithium extraction based on a rocking chair system of LiMn2O4/Li1–xMn2O4: self-driven plus external voltage driven. Sep. Purif. Technol. 259, 118154 (2021).
Xu, T. & Huang, C. Electrodialysis-based separation technologies: a critical review. AlChE J. 54, 3147–3159 (2008).
Brown, C. G. & Sherrington, L. G. Solvent extraction used in industrial separation of rare earths. J. Chem. Technol. Biotechnol. 29, 193–209 (1979).
El-Nadi, Y. A. Solvent extraction and its applications on ore processing and recovery of metals: classical approach. Sep. Purif. Rev. 46, 195–215 (2017).
Mooiman, M. B. The solvent extraction of precious metals: a review. In Proc. 17th International Precious Metals Conference 411–434 (International Precious Metals Institute, 1993).
Yordanov, A. T. & Roundhill, D. M. Solution extraction of transition and post-transition heavy and precious metals by chelate and macrocyclic ligands. Coord. Chem. Rev. 170, 93–124 (1998).
Mahandra, H., Faraji, F. & Ghahreman, A. Novel extraction process for gold recovery from thiosulfate solution using phosphonium ionic liquids. ACS Sustain. Chem. Eng. 9, 8179–8185 (2021).
Towler, G. & Sinnott, R. in Chemical Engineering Design 3rd edn (eds Towler, G. & Sinnott, R.) 631–733 (Butterworth-Heinemann, 2022).
Kislik, V. S. in Solvent Extraction (ed. Kislik, V. S.) 113–156 (Elsevier, 2012).
Vidal, O. in Mineral Resources and Energy (ed. Vidal, O.) 27–52 (Elsevier, 2018).
Sovacool, B. K. et al. Sustainable minerals and metals for a low-carbon future. Science 367, 30–33 (2020).
Butt, C. R. M. & Hough, R. M. Why gold is valuable. Elements 5, 277–280 (2009).
Murphy, K. Gold RRS 2022—Surge in recent discoveries. S&P Global Market Intelligence https://www.spglobal.com/marketintelligence/en/news-insights/research/gold-rrs-2022-surge-in-recent-discoveries (2022).
Calvo, G., Mudd, G., Valero, A. & Valero, A. Decreasing ore grades in global metallic mining: a theoretical issue or a global reality? Resources 5, 36 (2016).
Moreau, V., Dos Reis, P. C. & Vuille, F. Enough metals? Resource constraints to supply a fully renewable energy system. Resources 8, 29 (2019).
Cui, J. R. & Zhang, L. F. Metallurgical recovery of metals from electronic waste: a review. J. Hazard. Mater. 158, 228–256 (2008).
Zadra, J. B., Engel, A. L. & Heinen, H. J. Process for Recovering Gold and Silver from Activated Carbon by Leaching and Electrolysis (US Department of the Interior, Bureau of Mines, 1952).
Ford, P. et al. Economics of end-of-life materials recovery: a study of small appliances and computer devices in Portugal. Environ. Sci. Technol. 50, 4854–4862 (2016).
Beyuo, M. & Abaka-Wood, G. ZADRA elution circuit optimisation and operational experience at the CIL Plant of Gold Fields Ghana Limited. In Proc. 4th UMaT Biennial International Mining and Mineral Conference 161–167 (UMaTBIC, 2016).
Cutting cobalt. Nat. Energy 5, 825 (2020).
Babu, B. R., Parande, A. K. & Basha, C. A. Electrical and electronic waste: a global environmental problem. Waste Manag. Res. 25, 307–318 (2007).
Ding, Y. J. et al. Recovery of precious metals from electronic waste and spent catalysts: a review. Resour. Conserv. Recycl. 141, 284–298 (2019).
Yu, M. L., Wang, K. & Vredenburg, H. Insights into low-carbon hydrogen production methods: green, blue and aqua hydrogen. Int. J. Hydrog. Energy 46, 21261–21273 (2021).
Shestakova, M. & Sillanpää, M. Removal of dichloromethane from ground and wastewater: a review. Chemosphere 93, 1258–1267 (2013).
Priya, V. S. & Philip, L. Treatment of volatile organic compounds in pharmaceutical wastewater using submerged aerated biological filter. Chem. Eng. J. 266, 309–319 (2015).
Toth, A. J. et al. Novel method for the removal of organic halogens from process wastewaters enabling water reuse. Desalin. Water Treat. 130, 54–62 (2018).
Alder, C. M. et al. Updating and further expanding GSK’s solvent sustainability guide. Green Chem. 18, 3879–3890 (2016).
Larsen, C. et al. A tool for identifying green solvents for printed electronics. Nat. Commun. 12, 4510 (2021).
Lu, P. Y. & Alkire, R. C. Mass transfer in parallel plate electrolyzers with two‐phase liquid‐liquid flow. J. Electrochem. Soc. 131, 1059 (1984).
Alkire, R. & Köhler, J. Indirect electrochemical epoxidation of hexene in a liquid-liquid electrolyte. J. Appl. Electrochem. 18, 405–409 (1988).
Papageorgiou, N., Maier, W. F. & Grätzel, M. An iodine/triiodide reduction electrocatalyst for aqueous and organic media. J. Electrochem. Soc. 144, 876 (1997).
Masahiro, T., Akitsugu, O. & Taijiro, O. The chemical behavior of low valence sulfur compounds. VIII. The oxidation of sodium thiosulfate with ozone. Bull. Chem. Soc. Jpn 46, 3785–3789 (1973).
Rumble, J. R. CRC Handbook of Chemistry and Physics (CRC Press, 2017).
Sarla, M., Pandit, M., Tyagi, D. K. & Kapoor, J. C. Oxidation of cyanide in aqueous solution by chemical and photochemical process. J. Hazard. Mater. 116, 49–56 (2004).
Pei, Z. B. et al. Review of the I−/I3− redox chemistry in Zn-iodine redox flow batteries. Mater. Res. Bull. 141, 111347 (2021).
Li, X. et al. Symmetry-breaking design of an organic iron complex catholyte for a long cyclability aqueous organic redox flow battery. Nat. Energy 6, 873–881 (2021).
Orita, A., Verde, M. G., Sakai, M. & Meng, Y. S. A biomimetic redox flow battery based on flavin mononucleotide. Nat. Commun. 7, 13230 (2016).
Geankoplis, C. J., Hersel, A. A. & Lepek, D. H. Prentice Hall International Series in the Physical and Chemical Engineering Sciences (Prentice Hall, 2018).
Baker, D. A., East, G. C. & Mukhopadhyay, S. K. Synthesis and characterization of some disulfonyl azides as potential crosslinking agents for textile fibers. J. Appl. Polym. Sci. 79, 1092–1100 (2001).
Acknowledgements
This paper is based on work supported by the US Department of Energy, Office of Basic Energy Sciences under award no. DOE DE-SC0021409. This work is also partially supported by the NSF DMREF grant 2323988.
Author information
Authors and Affiliations
Contributions
S.R.C., A.F., J.E. and X.S. conceptualized and designed this work. S.R.C. and A.F. performed the experiments. S.R.C. and X.S. wrote the original paper. X.S. supervised the work and secured the funding. All authors analyzed the data and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors have applied for a provisional patent based on the technology proposed in the paper.
Peer review
Peer review information
Nature Chemical Engineering thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–38 and Discussion.
Source data
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cotty, S.R., Faniyan, A., Elbert, J. et al. Redox-mediated electrochemical liquid–liquid extraction for selective metal recovery. Nat Chem Eng 1, 281–292 (2024). https://doi.org/10.1038/s44286-024-00049-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44286-024-00049-x
This article is cited by
-
Electrochemical concentration pumping in liquid–liquid extractions
Nature Chemical Engineering (2024)
