
Abstract
Decarbonizing the steel industry, a major CO2 emitter, is crucial for achieving carbon neutrality. Escaping the grip of CO combustion methods, a key contributor to CO2 discharge, is a seemingly simple yet formidable challenge on the path to industry-wide net-zero carbon emissions. Here we suggest enzymatic CO hydration (enCOH) inspired by the biological Wood‒Ljungdahl pathway, enabling efficient CO2 fixation. By employing the highly efficient, inhibitor-robust CO dehydrogenase (ChCODH2) and formate dehydrogenase (MeFDH1), we achieved spontaneous enCOH to convert industrial off-gases into formate with 100% selectivity. This process operates seamlessly under mild conditions (room temperature, neutral pH), regardless of the CO/CO2 ratio. Notably, the direct utilization of flue gas without pretreatment yielded various formate salts, including ammonium formate, at concentrations nearing two molar. Operating a 10-liter-scale immobilized enzyme reactor feeding live off-gas at a steel mill resulted in the production of high-purity formate powder after facile purification, thus demonstrating the potential for decarbonizing the steel industry.

Similar content being viewed by others

Main
The steel/iron industry (7% of global CO2 emissions)1 is one of the primary contributors to greenhouse-gas emissions in the manufacturing field, along with electricity generation and transportation2,3. With the increasing demand for steel and iron predicted to continue, substantial reductions in CO2 emissions are imperative (annually 3 GtCO2 emission by 2050)2,4,5. A major portion of these emissions originates from the combustion of CO, a by-product of iron production, releasing two tons of CO2 per ton of manufactured steel2,4,6. Because of the toxicity of CO, its current method of disposal through combustion results in considerable CO2 discharge, exacerbating emissions. This reliance on combustion also foregoes the potential to utilize CO as a feedstock for synthesizing alternative fuels and chemicals, thereby missing a critical opportunity to replace fossil resources with more sustainable options. Hence, the capture or transformation of CO into value-added compounds without resorting to combustion is emerging as a prospective solution towards industry-wide decarbonization and, by implication, a substantial reduction in CO2 emissions.
The conversion of CO gas into commodity chemicals can be facilitated by metal-catalyzed chemical processes7,8. Among the two prevalent reactions, Fischer‒Tropsch synthesis performed on syngas streams has emerged as a possible conduit for converting CO from the steel and iron industry9,10, as opposed to the CO2-generating water‒gas‒shift reaction. Nevertheless, these processes require harsh operational conditions and high energy inputs/capital costs, in addition to requiring high-purity CO gas. In contrast, bioconversion of CO is performed under milder conditions, thus demanding potentially lower energy input. Several pioneering advancements have demonstrated the efficacy of biocatalytic CO utilization, including LanzaTech’s ethanol fermentation and the Muller group’s formate production via H2-dependent CO2 reductase (HDCR), both of which are derived from the acetogenic Wood‒Ljungdahl (WL) pathway11,12. LanzaTech’s process efficiently transforms low-cost waste gases into chemicals like ethanol, acetone and isopropanol using engineered acetogens, while the HDCR method uniquely converts CO2 to formic acid using H2 and electron transfer. The WL pathway, with its linear progression from CO2 to formic acid (eastern branch) and CO (western branch)13,14, presents a simpler implementation route compared to intricate circular CO2 fixation cycles, such as the Calvin Benson Bassham cycle (involving multiple enzymes and reactions; Supplementary Fig. 1)15. Its application appears particularly promising for sulfur-containing off-gases, as the WL pathway’s enzymes have evolved in sulfur-rich environments16,17. Despite these promising strides, obstacles still remain. For example, oxygen and toxic substances such as cyanide can impair stability, and the H2-dependent carbon fixation yield for complete carbon utilization poses further challenges. Although HDCR represents a promising system for CO2 conversion, it demands a continuous hydrogen supply to transform CO2 effectively. Bioconversion of industrial off-gas necessitates addressing issues such as inhibition by gas impurities (CN‒, SOx, NOx and O2), lower carbon yields and slower conversion rates at lower CO concentrations15,18,19,20,21,22,23,24,25.
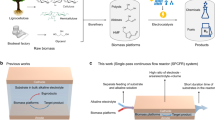
To address these challenges, we suggest an engineered in vitro enzymatic reaction inspired by the reverse western branch of the WL pathway to efficiently exploit industrial CO off-gas containing varied gas components and toxic impurities (Fig. 1a):

a, Conceptual overview of in vitro enCOH. This is inspired by the integration of the reverse western and eastern branches of the microbial WL pathway. b, Scheme of enCOH. CO is oxidized to CO2 by CODH, and CO2 is reduced to formate by FDH. The electron mediator (Md) transfers electrons between both enzymes. ACS, acetyl-CoA synthetase; Fd, ferredoxin; ox, oxidized; red, reduced.
The designed reaction demonstrates favorable thermodynamics, presenting a promising route for the direct utilization of real industrial off-gas. Importantly, our enzyme-catalyzed CO hydration (enCOH) reaction, formulated as CO + H2O → HCOOH (formic acid), occurs without any additional CO2 emission (Fig. 1b). The standard Gibbs free energy of the enCOH reaction, ΔG0′ = −17.4 kJ mol−1 at 25 °C and pH 7.0, further substantiates its thermodynamic feasibility (Supplementary Table 1). The toxic impurities typically found in flue gas (such as SOx, NOx, H2S and CN−) that usually deactivate metal catalysts and require complex and expensive pre-separation processes26,27,28,29 surprisingly exert negligible inhibition effects on either the CO dehydrogenase (CODH) or formate dehydrogenase (FDH) involved in the enCOH reaction (Supplementary Fig. 2), making it facilely applicable to direct industrial off-gas. Therefore, the enCOH in vitro system has potential due to its ease of control, reduced complexity, simplicity in terms of engineering and optimization, and flexibility under various conditions.
In this Article we report an enCOH reaction that is operational at atmospheric temperature and pressure, facilitating molar-scale formate production from industrial steel manufacturing off-gas and waste-plastic gasification. This process exhibits enduring stability in both laboratory settings and a semi-pilot-scale installation within a steel mill’s operational field.
Enzymatic hydration of CO to formate
The enCOH reaction initiates with CO as the starting substrate, leading to CO2 production through CO oxidation catalyzed by CODH. The subsequent spontaneous reaction catalyzed by FDH results in the generation of formic acid, with a theoretical yield of 1 mole of formic acid per 1 mole of CO (Fig. 1b). To ensure efficient enCOH, it is preferable that a complete cycle reaction take place through the regeneration of the electron mediator in the preceding one-pot reaction. Natural electron carriers (such as ferredoxins) are too specific and are also oxygen-sensitive30, so artificial options were examined for both CODHs and FDHs originated from various strains (Supplementary Table 2). Accordingly, the various viologens (methyl viologen (MV); benzyl viologen (BV); ethyl viologen (EV)) and a natural mediator (nicotinamide adenine dinucleotide (NAD)) were investigated based on their reaction energies, which were calculated based on the redox potential and standard Gibbs free energy at pH 7 (Supplementary Table 1 and Supplementary Fig. 3). These investigations suggested MV and EV to be more efficient mediators than BV or NAD, as they could supply enough energy to convert CO2 to formate.
In our enCOH in vitro system, we selected candidate enzymes for CODH and FDH considering key parameters such as reactivity, substrate affinity, protein productivity and the pH optimum (Supplementary Table 3): Carboxydothermus hydrogenoformans (Ch)CODH231 was considered for its high CO-oxidizing activity, ChCODH432 for its high CO affinity, Thermococcus onnurineus NA1 (To)CODH33 for its broad pH range, Methylobacterium extorquens (Me)FDH123,34 for its high CO2-reducing activity, Rhodobacter capsulatus (Rc)FDH35,36 for its high CO2 affinity and Thiobacillus sp. KNK65MA (Ts)FDH37,38 for its high expression and efficiency. Acetogen-derived CODH was excluded due to its low CO2-reducing activity in Escherichia coli (<0.04 U mg−1 for both NADH and EVred; 0.14 U mg−1 from the Müller group39). Unlike CODH, which mostly has a negligible backward reaction, for FDH we considered the relative rates \(\left(k_{{\mathrm{cat}},{\mathrm{C{O}}_2}}/k_{{\mathrm{cat}},{\mathrm{HCOO}}^{-}}\right)\) of the forward (CO2 reduction) and backward (formate oxidation) reactions (Supplementary Table 4). Furthermore, kinetic stability and oxygen sensitivity were addressed through immobilization and enzyme engineering29,40.
In evaluating individual enzyme’s efficiencies toward mediators, we measured each enzyme’s catalytic properties with different mediators in a controlled environment (pH 6.5, 30 °C), as shown in Extended Data Table 1 and Supplementary Fig. 4. Among the three tested mediators, the CODHs displayed no activity for NAD+, and the FDHs showed very low activity for reduced BV, suggesting that the overall reaction rate of enCOH using these mediators would be slow. In contrast, the use of EV enabled CO-oxidizing activity in all CODHs tested and high CO2-reducing activities in two metalloenzyme FDHs (MeFDH1 and RcFDH), suggesting that EV is the most effective mediator. When using EV as an electron mediator, the highest CO-oxidizing and CO2-reducing activities were observed with ChCODH2 (290 U mg−1 using EVox as the electron acceptor) and MeFDH1 (66 U mg−1 using EVred as the electron donor), respectively, among the six candidate enzymes.
To confirm the functionality of our proposed enCOH system involving CODH and FDH and to select a suitable enzyme combination set through experiments, we compared formate production with three commercially available mediators in their oxidized forms (BVox, EVox and NAD+) due to their ease of handling compared with MV. Upon conducting reactions with 99.998% CO (vol/vol), we observed formate production for 48 h only when using mediator EV and MeFDH1 (Fig. 2a). Interestingly, the highest formate production was observed in the following order for CODH: ChCODH2 (40.2 mM), ToCODH (7.7 mM) and ChCODH4 (2.6 mM), when combined with MeFDH1. RcFDH devoid of the stabilizer KNO3, as well as TsFDH lacking metal in the active site, showed no formate production in any combination with CODHs and mediators. Accordingly, the combined activities of enzymes in the enCOH system synergistically led to high formate production. These findings suggest that ChCODH2 and MeFDH1 are the best-performing set among the tested enzymes for enCOH.

a, Selection of CODH and FDH for enCOH. ChCODH2, ChCODH4 and ToCODH were the candidate CODHs, and MeFDH1, RcFDH and TsFDH were the candidate FDHs. Using BVox, EVox and NAD+ as electron mediators, the formate (reverse triangles) productivity for each combination of CODHs and FDHs was determined over a 48 h period (data are presented as the mean ± s.d. of n = 3 replicates). b, Time course of the biotransformation through enCOH in a closed system. The percent of CO (circles) and CO2 (triangles) was measured by GC and the formate (reverse triangles) amount was measured by HPLC over time. The yield (%, diamonds) of enCOH was calculated by dividing ∆formate into ∆CO (data are presented as mean ± s.d. of n = 3 replicates). c, Tracking of enCOH using isotopes 13CO and D2O. 13CO gas (magenta) and D2O (cyan) were used for testing, and the molecular weight of the formate produced via enCOH was determined by mass spectrometry (n = 1 replicate). A total of 98.9% (vol/vol) of the carbon was 12C and 99.9 % (vol/vol) of hydrogen was 1H, leading most formate to have 45 g mol−1 of molar mass18,19.
A more in-depth examination of the yield and selectivity of the reaction involving ChCODH2 and MeFDH1 was carried out by conducting enzymatic conversion of the gas substrate CO to formate in closed serum bottles (Fig. 2b). The amounts of CO, CO2 and formate were measured for 36 h in a closed system to estimate the yield of CO to formate. We observed that the formate gradually increased as CO was consumed, and, after 30 h, all the CO had been converted to formate. In the initial 12 h of the reaction, the formate production exhibited a slightly lower yield until the CO2 accumulated to the level of ~1 mM. This can probably be attributed to MeFDH1’s modest CO2 affinity, at 3.9 mM (Supplementary Table 4). However, approaching the end point of the reaction, the consumed amount of CO was completely matched by the amount of produced formate, suggesting stoichiometric conversion between CO and formate (Fig. 2b and Supplementary Fig. 5). To verify whether the enzymatic reaction products of enCOH were genuinely a result of CO combining with water to generate formic acid, we examined the substrate and product masses using isotopes (13CO). Our isotopic labeling experiments confirmed that the HCOO− was derived from 13CO and D2O (Fig. 2c). The molar mass of H13COO− was found to be 45.98 g mol−1, comparable to that of the H12COO− (44.98 g mol−1) obtained from 12CO/H2O conditions. Similarly, the H2O/D2O experiment with the same ratio showed a molar mass of D12COO− of 45.98 g mol−1. These results demonstrate that the carbon and hydrogen atoms of formate originated from the CO and water, respectively.
enCOH conversion of industrial off-gases to formate
An efficient enCOH system requires the simultaneous catalysis of CO and CO2 substrates by the CODH and FDH enzymes, with both performing optimally. To achieve this, factors such as substrate and product inhibition, physicochemical conditions like pH, temperature and buffer, as well as the enzyme ratio, should be considered41. Interestingly, no inhibitory crosstalk was observed between the formate product on ChCODH2 and the CO substrate on MeFDH1 (Supplementary Fig. 6). This is probably due to the tungsten-containing active site of FDH being tolerant to CO42 and the size of formate hindering its passage through the substrate tunnel to access the nickel-containing active site of CODH (Supplementary Fig. 7). In addition to the inhibition effect, the pH profile revealed that both enzymes were active between pH 6.5 and 8, as shown in Supplementary Fig. 8a. A pH of 6.5 was chosen for enCOH because the overall reaction rate was governed by the enzymatic reaction of MeFDH1, which exhibited lower activity compared to ChCODH2 (Extended Data Table 1). In terms of temperature, the enzyme activity progressively increased within the 20‒70 °C range. However, based on kinetic stability, activity was better maintained at temperatures below 30 °C (Supplementary Fig. 8b‒d), suggesting a trade-off between activity and stability.
The optimal temperature range for cost-effective and sustainable enzyme use would be at a temperature where there is a balance between maintaining a reasonable level of enzyme activity and ensuring their stability for long-term use. For example, higher temperatures may offer greater initial activity but at the expense of reduced enzyme stability, leading to frequent replacement or maintenance costs. By contrast, lower temperatures may yield slightly less activity but with greater stability and longevity, reducing overall operational costs. Long term, therefore, it is deemed more cost-effective to sustain consistent activity at lower temperatures rather than having high activity at elevated temperatures. The impact of various buffers (sodium phosphate, bis-tris propane, imidazole and MOPS (3-(N-morpholino) propanesulfonic acid)) at 50 mM concentrations appeared minimal, but sodium phosphate buffer emerged as the most effective (Supplementary Fig. 8e). Sodium phosphate, due to its affordability and availability compared to MOPS and other buffers like bis-tris propane and imidazole, offers a cost-effective, easier to source and practical solution for process scale-up. This choice effectively aligns enzyme performance with the economic considerations essential for industrial use and commercialization. Furthermore, a higher enzyme ratio of ChCODH2 to FDH was shown to be beneficial, because a higher EVred/EVox ratio is preferred for CO2 reduction catalyzed by FDH (Supplementary Fig. 8f). Based on these findings, we concluded that both enzymes—ChCODH2 and MeFDH1—can function effectively together under ambient/neutral conditions.
In steel-mill flue-gas applications, it is essential for the two enCOH enzymes to be tolerant to potential inhibitors such as O2, HCN (0.3–1.5 g N−1 m−3) and SOx (0.1–0.8 g N−1 m−3), which are present in the flue gas of steel mills43. As shown in Supplementary Table 5, these gases exhibit variable oxygen concentrations (0‒17%, vol/vol), necessitating the consideration of oxygen tolerance in industrial enzyme applications. For O2, this challenge can be addressed by employing the recently developed O2-tolerant ChCODH2 A559W29,44 and the well-established O2-tolerant MeFDH123. Apart from this, when examining the inhibitory effect, both ChCODH2 and MeFDH1 retained their activities at aqueous cyanide and bisulfite concentrations corresponding to the range found in real flue gas (~0.05 µM cyanide; ~4 µM bisulfite). However, those enzyme activities declined when exposed to excessively high levels of cyanide and bisulfite, with half-maximal inhibitory concentration (IC50) values of 2 µM for ChCODH2 and 600 µM for MeFDH1 towards cyanide. FDH activity fully recovered post-inhibition, unlike the limited restoration of CODH. Such levels are typically not observed in steel-mill flue gases, which contain far lower concentrations (Supplementary Fig. 2). ChCODH2 undergoes competitive inhibition by cyanide due to the binding of cyanide to the Ni atom of the catalytic center (Ni‒Fe‒S cluster), whereas MeFDH1 exhibits greater tolerance to cyanide19. No definitive inhibition mechanism caused by bisulfite on the two enzymes has been identified, and resistance to bisulfite is considerably higher than that of cyanide.
Leveraging this resilience, the production of formate using an O2-tolerant enzyme-based enCOH (ChCODH2 A559W and MeFDH1)23,29 was evaluated in a closed reactor using real flue gas. Three representative waste gases, including coke-oven gas (COG), blast-furnace gas (BFG) and Linz‒Donawitz gas (LDG), bottled gases from a Hyundai steel mill in Korea (Supplementary Table 5) and waste gas from mixed plastics (solid recovered fuel (SRF)) from the gasifier of Korea Institute of Energy Research (KIER) were used for the enCOH reaction (Fig. 3a and Supplementary Table 6). A mass-balance analysis for CO consumption and formate production was conducted by measuring CO levels in the gas phase and formate concentrations in the aqueous phase. Despite variations in the CO content and complex gas compositions of the supplied gases, all CO molecules were converted to formate, corroborating the theoretical yield of the enCOH reaction. As expected, LDG with higher carbon content produced a higher absolute amount of formate (~460 µmol) relative to the CO input, and COG with the lowest carbon content resulted in less formate (~93 µmol). Interestingly, COG, BFG, LDG and SRF-derived gas all achieved 100% conversion yields. This observation supports that an inexpensive LDG is a suitable waste gas for conversion into fuels and chemicals, due to the high content of recyclable CO/CO2. Based on the gas composition of CO/CO2, we classified waste real gas into three categories: COG/BFG, equivalent ratios; LDG, high CO/low CO2; SRF-derived gas, low CO/high CO2. As shown in Fig. 3b, enCOH transformed the low CO content in waste-plastic-derived gas into formate. These findings demonstrate that enCOH can effectively utilize CO across diverse gas compositions because of its high substrate affinity for CO29. COG, BFG, LDG and SRF-derived gas have various compositions of gaseous components other than CO/CO2, but these had no significant adverse effects on the enzymatic conversion of enCOH. For example, COG has a high H2 content29,45, but this had no impact (Fig. 3a‒b and Supplementary Fig. 9). Based on this, it is expected that enCOH can utilize real waste gas with various compositions.

a, Influence of using real flue gas (BFG, COG, LDG) with enCOH. Industrial flue gases were used in enCOH. CO consumption (circles), CO2 (triangles) and formate (inverse triangles) production, as well as yield (%, diamonds) were measured over time (data are presented as the mean ± s.d. of n = 3 replicates). b, Influence of using plastic-derived gas on enCOH. The gas derived from SRF gasification was used in enCOH. CO, CO2 and formate were observed (data are presented as the mean ± s.d. of n = 3 replicates). The icon for gasification was made by Freepik from Flaticon (www.flaticon.com).
To achieve cost-effective enzymatic formate production, the activity of enCOH enzymes must be stably maintained during the conversion of CO to formate when LDG gas with higher CO/CO2 is applied for long-term operation. A repeated reuse experiment was conducted, immobilizing CODH and FDH on nickel–nitrilotriacetic acid (Ni-NTA) resin (Fig. 4a,b). Consequently, formate productivity was sustained without sacrificing enzyme activity for over ten reuse cycles. This suggests that stable chemical production is attainable through enCOH using real flue gas due to the enzymes’ resilience to toxic inhibitors such as cyanide and bisulfite.

a, Schematic of the experimental procedure. Enzymes with a His6-tag were immobilized on Ni-NTA resin for ten cycles of reuse, with no severe loss in conversion rate. The reaction was performed at room temperature (rt) using a rubber-sealed serum bottle (115 ml) with LDG-purged reaction buffer (20 mM EVox, 200 mM sodium phosphate buffer pH 6.5 with purged LDG gas for 1 h). The immobilized enzyme was subsequently recovered through a disposable open column, washed twice with 200 mM sodium phosphate buffer pH 6.5 and reused after each reaction. b, Repeated reuse of enzymes for enCOH with LDG. By using CODH and FDH, immobilized on Ni-NTA agarose beads, the reusability of the enzymes was tested. CO (circles) consumption, CO2 (triangles) and formate (reverse triangles) production, and yield (%, diamonds) were measured every 12 h (n = 1 replicate). Shaded bands denote the ten cycles.
Scaling-up enCOH for steel-mill field implementation
To explore the applicability of the enCOH system for continuous-flow reactions, a customized 100-ml enzyme reactor was designed and tested (Fig. 5a and Supplementary Fig. 10). Because formic acid production led to a continuous pH drop due to the low pKa value (pKa = 3.745), a neutralizing agent was required to keep the pH constant. We operated a 100-ml-scale reactor for enCOH under pH 6.5 control by continuously purging LDG, and evaluated the productivity and efficiency of the enCOH reaction using three representative neutralizers: NaOH, KOH and NH4OH (Fig. 5b). For all the neutralizers, formate production remained stable for 64 h, yielding ~1.1 M (for NaOH), 1.2 M (for KOH) or 1.8 M (for NH4OH). The resulting formate salts—sodium (HCOONa, 88.8% purity), potassium (HCOOK, 94.6% purity) and ammonium (HCOONH4, 92.1% purity)—were easily obtained (Fig. 5c and Supplementary Fig. 11). Throughout this process, the use of NH4OH not only yielded high productivity, but also generated ammonium formate, a notable compound that is challenging to produce via conventional industrial processes46,47 and is synthesized easily through enzymatic reactions. Applying a catalyst to formate salts is anticipated to facilitate efficient hydrogen production, positioning them as promising hydrogen carriers48, because formate salts are more eco-friendly and safe than formic acid, offering zero CO2 emissions, reversible reactions, favorable thermodynamics and stable storage with fewer requirements.

a, Long-term operation with continuous gas purging (LDG) and pH control in a 100-ml-scale reactor using various types of pH neutralizer. The 3D structures (CO, CO2, formate and EV) were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov). b, The enCOH produced formate in the continuous gas flow system using NaOH, KOH or NH4OH as the pH neutralizer (data are presented as the mean ± s.d. of n = 3 replicates). c, Extraction of three formate salts in a 100 ml reactor. The produced formate was purified to sodium, potassium or ammonium formate powder. In hydrogen production, formate salts undergo dehydrogenation, releasing hydrogen and transforming into bicarbonate. The icon was made by Freepik from Flaticon (www.flaticon.com). d, Feasibility evaluation of the biocatalytic system for formate production when utilizing synthetic gases or industrial off-gases. In commercial-scale operations, LanzaTech’s ethanol production reaches up to 10 g l−1 h−1. Due to the limited available details on catalyst usage and titer, direct comparisons are challenging. Nonetheless, this system’s ethanol productivity is recognized as commercially viable.
To evaluate the biocatalytic efficiency of the enCOH system, we compared it with recent bioconversion technologies employed for generating value-added products from COx gaseous sources (Fig. 5d and Supplementary Table 7). The enCOH system enables the production of high formate concentrations and notable productivity even using real flue gas; however, there are few reported works involving testing with real off-gases. In the context of production performance, the enCOH system demonstrates the following: (1) unlike whole-cell catalysis, this in vitro enzymatic system effectively overcomes challenges such as cell disruption and by-product formation by means of a combination of efficient enzymes, thus achieving high titers, and (2) leveraging CODH and FDH coupling, this system attains high productivity without external energy, distinguishing it from methods such as electrocatalysis and photocatalysis. This shows the in vitro enzyme system’s ability to simultaneously achieve high titer and productivity, making it a competitive and viable option for industrial-scale production. This highlights the potential utility of the enCOH system for industrial applications.
Finally, a scaled-up 10-l reactor was designed for field operation of the enCOH reaction (Fig. 6a and Supplementary Fig. 12). The pilot reactor was installed to utilize a live feed of LDG directly connected from Hyundai Steel’s site (Dangjin, Korea), without energy input. We observed that ~298.5 g of formate was produced during the 69.5-h enCOH reaction under ambient conditions (Fig. 6b). We also demonstrated repeated reuse of the enCOH biocatalysts (Fig. 6c). Unexpectedly, a temporary gas-supply interruption (at 25 h) and low temperature in the actual field environment negatively affected formate productivity (an electric temperature-control system could not be installed due to the strict safety regulations at the steel mill). Nonetheless, the enCOH system’s stress test results indicate that actual waste by-product gas can be effectively used to produce the valuable chemical formate. Given the system’s minimal susceptibility to variations in the CO content, catalytic inhibitors and gas composition, the enCOH reaction is a promising way to transform real flue gas into valuable products in industrial applications.

a, A scheme of the 10-l enzyme reactor (with a continuous feed of live LDG). enCOH was conducted in a continuous gas flow and pH-controlled system with immobilized enzymes in a large-scale reactor. b, Field operation of the enCOH reactor (10 l) at Hyundai Steel, with a continuous live LDG feed (n = 1 replicate). c, Repeated reuse of the enCOH biocatalyst for three cycles (n = 1 replicate). Shaded bands indicate the three cycles.
Discussion
Our technology facilitates the transformation of flue CO into formate at a near two-molar scale, demonstrating enzyme engineering’s potent role in the crafting of practical industrial biocatalysts. Formate, being a versatile precursor, is increasingly a target in CO2 reduction, chemical synthesis and energy production12,49,50. For green hydrogen, formate provides safe liquid storage49. We can explore ethyl/methyl formate and its use as a carbon source for chemicals such as polyhydroxyalkanoates45, using natural and metabolic engineered strains50. Its versatility positions formate as a key player in reducing environmental footprints and advancing sustainable practices across industries. This oxygen-tolerant enzymatic technology29,40 has shown potential for efficient mixed-waste gas conversion into valuable formate, advancing industrial CO2 reduction and sustainable hydrogen carrier production.
In the pilot experiments, we connected directly to a live feed of LDG at Hyundai Steel, providing a realistic simulation of steel-mill conditions. This approach revealed how the enCOH system might perform in actual industrial settings. The live feed set-up brought unique operational challenges, such as stringent electricity usage constraints. Importantly, using a live LDG feed allowed us to examine the interactions of enzymes with potential contaminants as well as the reaction consistency, offering critical insights for practical application. The implementation of a 10-l-scale enzyme reactor at a steel company is an important step forward, demonstrating its potential for scalability and integration into industrial processes. Major steel companies in South Korea (POSCO and Hyundai Steel) are now exploring opportunities for incorporating this technology into their processes. This not only emphasizes the versatility and applicability of enzyme engineering but also its potential to promote sustainability within industrial processes20,24.
In conclusion, the enzymatic molar-scale production of formate from complex industrial off-gases using a rationally designed in vitro reaction is a notable stride towards sustainable energy production systems and the realm of industrial biotechnology. The potential ramifications of this technology are considerable, providing invaluable insights for scientific, environmental and industrial sectors seeking to reduce greenhouse-gas emissions and support sustainable industrial processes.
Methods
Gene synthesis and cloning for expression
All the ChCODH2 (UniProt accession number Q9F8A8) and ChCODH4 (Q9F8A8) genes from C. hydrogenoformans29 were synthesized by Macrogen (Korea), optimized for NdeI and BamHI restriction sites, and cloned into pET-28a(+) (Novagen), which encodes an N-terminal His-tag. These constructs were then transformed into E. coli BL21 harboring pRKISC51 for expression. The tungsten-based MeFDH1 genes, fdh1A (C5ATT7) and fdh1B (C5ATT6), were integrated into pCM110 and expressed in M. extorquens AM134, because expression in E. coli was unsuccessful. The molybdenum-based RcFDH genes (fdhABC, D5AQH0‒2; fdhE, D5AQG8) from R. capsulatus35,36 were designed for NheI and SacI restriction sites, cloned into pTrcHis A, and expressed in E. coli MC1061. The non-metallic TsFDH gene (Q76EB7) from Thiobacillus sp. KNK65MA, optimized for NheI and EcoRI restriction sites, was integrated into pUC57 and expressed in E. coli BL2137. Finally, the ToCODH gene (B6YWP3) from T. onnurineus NA1 was optimized for NcoI and NotI restriction sites, synthesized, and confirmed by sequence verification prior to cloning33. The related gene, plasmid and host details are provided in Supplementary Table 8.
Enzyme expression and purification
Except for MeFDH1, the enzymes utilized in this study were acquired via heterologous expression in E. coli. All enzymes were isolated using Ni-NTA agarose beads to purify His6-tagged proteins under anaerobic conditions. The expression and purification process, primarily focusing on the enCOH enzymes ChCODH2 and MeFDH1, is described here, aligning with methods reported in previous research29,30,33,34,35,36,37.
Escherichia coli cultures expressing ChCODH2, ChCODH4 and ToCODH were grown in modified terrific broth medium supplemented with 0.02 mM NiCl2, 0.1 mM FeSO4, 2 mM l-cysteine and antibiotics (kanamycin 50 μg ml−1, tetracycline 10 μg ml−1), at 37 °C and 200 rpm. Upon reaching an optical density at 600 nm (OD600) of between 0.4 and 0.6, overexpression of CODH was induced with 0.2 mM isopropyl β-d-1-thiogalactopyranoside (IPTG), 0.5 mM NiCl2, 1 mM FeSO4 and 50 mM KNO3, after 1 h of N2 flushing at 30 °C. For MeFDH1 expression52, the defined medium with 16 g l−1 succinate as a carbon source and 9.9 mg l−1 Na2WO4 was used, with M. extorquens AM1, at 30 °C and 200 r.p.m. At OD600 0.4–0.6, 0.5% (vol/vol) methanol was added for induction. RcFDH and TsFDH were expressed in LB medium under conditions aligning with reported methodologies36,37. RcFDH expression required 150 μg ml−1 ampicillin, 1 mM sodium molybdate and 20 µM IPTG, incubated at 30 °C with 130 rpm agitation. TsFDH expression employed 50 μg ml−1 ampicillin and 1 mM IPTG, incubated at 37 °C and agitated at 200 rpm.
For kinetic analysis, cells were lysed either by sonication in an anaerobic chamber (COY Laboratory Products) or by a homogenizer (GEA Mechanical Equipment) for larger enCOH reaction preparations (100 ml reactor). The lysis buffers varied depending on the enzyme: CODH-expressing cells utilized a 50 mM sodium phosphate buffer (pH 8.0) of 300 mM NaCl, 2 mM dithioerythritol (DTE) and 2 µM resazurin; FDH-expressing cells employed a 50 mM MOPS (3-(N-morpholino)propanesulfonic acid) buffer (pH 7.0) containing 300 mM NaCl, 2 mM DTE and 2 µM resazurin. After lysis, the supernatant from the centrifugation (14,000g, 4 °C, 20 min) was mixed with Ni-NTA agarose beads and incubated for 15 min in an anaerobic chamber. The enzyme-bound beads were washed and directly used for enCOH reactions or eluted for further assays. The elution buffers were similar to the lysis buffers but contained 20‒250 mM imidazole for CODH and 20‒300 mM imidazole for FDH. An additional 10 mM KNO3 was used for RcFDH purification.
Enzyme activity measurement
For the CODH activity assay, a 200 mM sodium phosphate reaction buffer (pH 6.5) containing 20 mM oxidized mediators (BVox; EVox; NAD+) and 2 mM DTE was saturated with 99.998% (vol/vol) CO gas (Air Liquide Korea). The reaction was initiated with 0.1 µg of enzyme, and the activity was estimated at 30 °C by spectrophotometrically monitoring the initial rate of absorbance increase at appropriate wavelengths for each reduced mediator (BVred, ɛ578 = 9,750 M−1 cm−1; EVred, ɛ546 = 10,000 M−1 cm−1; NADH, ɛ340 = 6,220 M−1 cm−1)29,30,53.
For the FDH activity assay, the reaction was initiated by adding 1 μg of enzyme to a 200 mM sodium phosphate buffer (pH 6.5) containing 100 mM sodium bicarbonate, 0.1 mM reduced mediators (BVred, EVred, NADH) and 2 mM DTE at 30 °C. Activity was estimated by tracking the initial rate of decline in absorbance for each reduced mediator spectrophotometrically. Reduced viologens, BVred and EVred, were prepared anaerobically by mixing 200 mM oxidized viologens with 1 g of zinc and filtering through a 0.45-µm syringe filter54. All experiments were performed in triplicate.
Enzyme catalytic properties
Kinetic analysis of enzyme activity was performed using varying concentrations of electron mediators (0.5‒8 mM for CODH-mediated CO oxidation and 0.006‒0.1 mM for FDH-mediated CO2 reduction) in conditions identical to the activity assays. The kinetic parameters for BV, EV and NAD(H) were determined using Hanes‒Woolf plots. Enzyme activity profiles across different pH values (6‒8) and temperatures (20‒70 °C) were assessed using pH-adjusted buffers and 200 mM sodium phosphate buffer (pH 6.5), respectively.
Inhibition of CODH activity by formate was evaluated using a CO-saturated sodium phosphate buffer (pH 6.5) with 20 mM EVox, 2 mM DTE, 2 µM resazurin and varying formate concentrations (0‒1 M). The inhibitory effect of CO on FDH activity was assayed in a sodium phosphate buffer (pH 6.5) supplemented with 100 mM sodium bicarbonate, 0.1 mM EVred, 2 mM DTE, 2 µM resazurin and different CO concentrations (0‒1 mM). According to Henry’s law, a 99.998% (vol/vol) CO purged buffer at room temperature and pressure contains ~1 mM CO. To assess enzyme inhibition by KCN and NaHSO3, the enzymes (4 µg) were anaerobically incubated with varying concentrations of inhibitors (10 nM–20 mM) for 30 min in sodium phosphate buffer (pH 6.5). The residual enzyme activities were subsequently measured. In the recovery experiments for enzymes against KCN and sodium bisulfite inhibitors, Ni-NTA-immobilized CODH and FDH were treated with 10 mM inhibitors in a time-course experiment spanning 2 h. After the treatment period, the enzymes were washed twice with 50 mM bis-tris-propane buffer (pH 6.5). Residual activity was subsequently measured over time using 20 mM EVox for CO oxidation and 0.1 mM EVred for CO2 reduction. All experiments were performed in triplicate.
High-performance liquid chromatography and gas chromatography analysis
In this study, formate (reaction product) was analyzed using high-performance liquid chromatography (HPLC), and the gaseous components, including CO (substrate), CO2 (intermediate) and other gases such as H2 and N2, were quantified via gas chromatography (GC) to monitor their changes during the enzymatic reactions. Immediately after sampling, formate from the enCOH reaction solution was quenched using a 6 M H2SO4 solution in a 10:1 ratio. HPLC measurements utilized an Aminex HPX-87H column (Bio-Rad) and an RID-20A refractive index detector (Shimadzu) (mobile phase, 5 mM H2SO4; flow rate, 0.6 ml min−1).
The gas content in the headspace of the serum bottle from enCOH experiments was analyzed using a 7890B GC-TCD system (Agilent Technologies). A 0.5-ml gas sample was obtained using a gas-tight syringe (Trajan Scientific and Medical). GC analyses employed a Carboxen-1000 column (Supelco) with argon as the carrier gas (flow rate, 30 ml min−1). The oven temperature was initially set at 35 °C for 5 min, and then ramped to 225 °C at a rate of 20 °C min−1. All experiments were performed in triplicate.
Electrospray ionization mass spectrometry, Fourier-transform infrared spectroscopy and NMR analysis
The isotopic identity of enCOH products was confirmed using electrospray ionization mass spectrometry (ESI-MS). Reaction mixtures, prepared with 13CO (99% atom 13C, Cambridge Isotope Laboratories) or D2O (99.9% atom D, Sigma-Aldrich), yielded formates with distinct masses. Either 12CO or 13CO was introduced into a reaction buffer (10 mM EVox, 2 mM DTE, 2 µM resazurin; for D2O experiments, up to 50% (vol/vol) D2O) in a serum bottle. Reactions were initiated by injecting 2,000 U ChCODH2 and 20 U MeFDH1, and quenched after 24 h with 10 M KOH for ESI-MS analysis. The ESI-MS set-up (JMS-T100LP, JEOL) was operated in negative mode, with a needle voltage of −2,000 V, a 250 °C desolvation chamber, −30 V and 80 °C orifice 1, −5 V ring lens, −10 V orifice 2 and 100 V ion guide RF. A survey time-of-flight MS experiment scanned m/z from 10 to 100 for isotope-labeled formate.
To characterize the formate salts produced from enCOH reactions, Fourier-transform infrared spectroscopy (FT-IR) and NMR were used. For three formate salts (sodium, potassium and ammonium) analysis, FT-IR was used. Those salts were produced with immobilized enzymes (35 U of Ni-NTA immobilized ChCODH2 A559W and 125 U of Ni-NTA MeFDH1) in reaction buffer with 10 mM EVox, 2 mM DTE and 2 µM resazurin. FT-IR spectra were acquired on a Nicolet iS50 spectrometer (Thermo Fisher Scientific) in attenuated total reflection mode. 1H-NMR spectra were acquired using a Bruker 400-MHz AVANCE III HD spectrometer at ambient temperature, with D2O as the reference (D2O, δ = 4.69 ppm; formate, δ = 8.37 ppm).
Evaluation of enCOH enzyme candidates
To identify the most effective enzymes for the enCOH reaction, combinations of various enzymes and electron mediators were examined. For CODHs, 100 µg each of ChCODH2, ChCODH4 and ToCODH were tested. For FDHs, 250 µg each of MeFDH1, RcFDH and TsFDH were evaluated. Serum bottles containing a 200 mM bis-tris propane buffer (pH 6.5) supplemented with 10 mM electron mediators (BVox, EVox and NAD+), 2 mM DTE and 2 µM resazurin were purged with 99.998% (vol/vol) CO gas for 1 h. Reactions at room temperature were initiated by enzyme injection, and formate concentration was measured after 48 h. For the time-course analysis, the conditions remained the same, employing ChCODH2, MeFDH1 and EVox. The reaction was monitored over time using both HPLC and GC. All experiments were performed in triplicate.
Enzymatic reaction in sealed serum bottles
The enCOH reactions were performed at room temperature in 120-ml serum bottles (sealed with rubber) containing 200 mM sodium phosphate buffer (pH 6.5), 10 mM EVox, 2 mM DTE and 2 µM resazurin. The bottles were purged for 1 h with test gases (99.998% (vol/vol) CO or real flue gases, including COG, BFG, LDG or SRF-derived gas) to fill the headspace. COG, BFG and LDG (compressed to 4‒5 MPa in 47-l cylinders) were sourced from Hyundai Steel29,30, and SRF-derived gas (compressed to 9 bar in 10-l cylinders) were from the Korea Institute of Energy Research30,55,56. The reactions were initiated by injecting 35 U of ChCODH2 A559W mutant and 125 U of MeFDH1, then the bottles were shaken at 150 rpm at room temperature. The pressure was equilibrated at each sampling by supplying N2 gas (99.999%, vol/vol) to compensate for the gas molecule consumption. All experiments were performed in triplicate.
For repeated enzyme usage, enzymes immobilized on Ni-NTA agarose beads were employed. The reactions contained 200 mM sodium phosphate buffer (pH 6.5), 10 mM EVox, 2 mM DTE, 2 µM resazurin, 35 U of immobilized ChCODH2 A559W mutant and 125 U of immobilized MeFDH1. Post-reaction beads were collected in a disposable column and washed twice with 20 ml of 200 mM sodium phosphate buffer (pH 6.5). LDG was supplied every 12 h after replenishing with fresh sodium phosphate buffer solution (pH 6.5) containing 10 mM EVox, 2 mM DTE and 2 µM resazurin.
Enzymatic reaction in the 100-ml bubble column reactor
To optimize gas substrate reactivity in the enzymatic reactions, we utilized a custom-designed 100-ml bubble-type column reactor, fabricated by newhillbiotech. This reactor is a four-necked glass cylinder with a bottom-up gas-supply system, a diameter of 29 mm and height of 563 mm.
The enCOH reactions were conducted at room temperature within this reactor, which was filled with 50 mM sodium phosphate buffer (pH 6.5), 10 mM EV, 2 mM DTE, 35 U of immobilized ChCODH2 A559W mutant and 125 U of immobilized MeFDH1. The reactor was continuously supplied with real flue LDG (compressed to 4‒5 MPa in a 47-l cylinder) at a rate of 100 ml min−1, introduced via a sintered glass filter installed at the bottom of the reactor. To maintain a pH of 6.5 in each experiment set and generate distinct types of formate salt, we utilized neutralizers including 10 M solutions of NaOH, KOH and NH4OH. All experiments were performed in triplicate.
10-l enzymatic reactor installation at Hyundai steel
A 15-l bubble column reactor (STS 316L; diameter, 16.5 cm; height, 80.6 cm) was customized and constructed by CNS Engineering. Key details of the reactor design are presented in Supplementary Fig. 12. The reactor was operated at Hyundai Steel using ChCODH2 A559W and MeFDH1 proteins, expressed in a customized fermenter by culturing CODH-expressing E. coli and FDH-expressing M. extorquens cells. Summary data, including the total weight of cultured strains, the total amount of expressed proteins and protein activities, are provided in Supplementary Table 9 and Supplementary Fig. 13. For large-scale protein production, ChCODH2 A559W and MeFDH1 were expressed under anaerobic and aerobic conditions, respectively. This process took place in 5-l (using the dual fermenter system MARADO-05D-PS) and 100-l (utilizing a customized pilot-scale fermentation system) fermenters, both constructed by CNS Engineering. A video demonstrating the set-up and operation of the reaction apparatus is provided in Supplementary Video 1. During operation, LDG from the steel mill was continuously supplied via a compressor (compressed to 3 bar) into the 15-l reactor, which held 10 l of 50 mM sodium phosphate solution (pH 6.5), supplemented with 1 mM EVox, 2 mM DTE, 3.5 kU of immobilized ChCODH2 A559W and 12.5 kU of immobilized MeFDH1. The pH was maintained at 6.5 by adding 10 N NaOH as a neutralizing agent.
Salt formate separation
Following the 100-ml reactor operation, the solution was filtered through a sintered glass filter, followed by an activated carbon bed (10 g, Darco-G60, Sigma-Aldrich). The adsorptive properties of activated carbon enabled formate separation, based on the reported affinity of viologens for activated carbon57,58. The recovered solution was dried in a convection oven at 90 °C overnight to yield powdered solids. The purity and identity of the produced formate were verified using HPLC, FT-IR and NMR analyses, respectively (Supplementary Table 10).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this Article.
Data availability
All data supporting the findings of this study are available within the article and the Supplementary Information. All source data have been deposited on figshare at https://doi.org/10.6084/m9.figshare.25433956.v2 (ref. 59). 3D chemical structures data for CO, CO2, formate and ethyl viologen are available in the PubChem database (https://pubchem.ncbi.nlm.nih.gov/).
References
Iron and Steel Technology Roadmap: Towards More Sustainable Steelmaking (International Energy Agency, 2020). https://www.iea.org/reports/iron-and-steel-technology-roadmap
Kohse-Hoeinghaus, K. Combustion, chemistry and carbon neutrality. Chem. Rev. 123, 5139–5219 (2023).
Yang, F., Meerman, J. C. & Faaij, A. P. C. Carbon capture and biomass in industry: a techno-economic analysis and comparison of negative emission options. Renew. Sust. Energ. Rev. 144, 111028 (2021).
De Ras, K., Van de Vijver, R., Galvita, V. V., Marin, G. B. & Van Geem, K. M. Carbon capture and utilization in the steel industry: challenges and opportunities for chemical engineering. Curr. Opin. Chem. Eng. 26, 81–87 (2019).
Sun, Y. Q. et al. Decarbonising the iron and steel sector for a 2 °C target using inherent waste streams. Nat. Commun. 13, 297 (2022).
Wang, P. et al. Efficiency stagnation in global steel production urges joint supply- and demand-side mitigation efforts. Nat. Commun. 12, 2066 (2021).
Khodakov, A. Y., Chu, W. & Fongarland, P. Advances in the development of novel cobalt Fischer–Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuels. Chem. Rev. 107, 1692–1744 (2007).
Gokhale, A. A., Dumesic, J. A. & Mavrikakis, M. On the mechanism of low-temperature water gas shift reaction on copper. J. Am. Chem. Soc. 130, 1402–1414 (2008).
Santos, V. P. et al. Metal organic framework-mediated synthesis of highly active and stable Fischer–Tropsch catalysts. Nat. Commun. 6, 6451 (2015).
Liu, B. et al. CO2 formation mechanism in Fischer–Tropsch synthesis over iron-based catalysts: a combined experimental and theoretical study. Catal. Sci. Technol. 8, 5288–5301 (2018).
Köpke, M., Mihalcea, C., Bromley, J. C. & Simpson, S. D. Fermentative production of ethanol from carbon monoxide. Curr. Opin. Biotechnol. 22, 320–325 (2011).
Schuchmann, K. & Muller, V. Direct and reversible hydrogenation of CO2 to formate by a bacterial carbon dioxide reductase. Science 342, 1382–1385 (2013).
Ljungdahl, L. G. & Wood, H. G. Total synthesis of acetate from CO2 by heterotrophic bacteria. Annu. Rev. Microbiol. 23, 515–538 (1969).
Ragsdale, S. W. & Pierce, E. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 1784, 1873–1898 (2008).
Fast, A. G. & Papoutsakis, E. T. Stoichiometric and energetic analyses of non-photosynthetic CO2-fixation pathways to support synthetic biology strategies for production of fuels and chemicals. Curr. Opin. Chem. Eng. 1, 380–395 (2012).
Martin, W. F., Sousa, F. L. & Lane, N. Energy at life’s origin. Science 344, 1092–1093 (2014).
Nisbet, E. G. & Sleep, N. H. The habitat and nature of early life. Nature 409, 1083–1091 (2001).
Jeoung, J. H. & Dobbek, H. Structural basis of cyanide inhibition of Ni, Fe-containing carbon monoxide dehydrogenase. J. Am. Chem. Soc. 131, 9922–9923 (2009).
Ha, S. W. et al. Interaction of potassium cyanide with the [Ni–4Fe–5S] active site cluster of CO dehydrogenase from Carboxydothermus hydrogenoformans. J. Biol. Chem. 282, 10639–10646 (2007).
Can, M., Armstrong, F. A. & Ragsdale, S. W. Structure, function and mechanism of the nickel metalloenzymes, CO dehydrogenase and acetyl-CoA synthase. Chem. Rev. 114, 4149–4174 (2014).
Rivas, M. G., Gonzalez, P. J., Brondino, C. D., Moura, J. J. G. & Moura, I. EPR characterization of the molybdenum(V) forms of formate dehydrogenase from Desulfovibrio desulfuricans ATCC 27774 upon formate reduction. J. Inorg. Biochem. 101, 1617–1622 (2007).
Robinson, W. E., Bassegoda, A., Reisner, E. & Hirst, J. Oxidation-state-dependent binding properties of the active site in a Mo-containing formate dehydrogenase. J. Am. Chem. Soc. 139, 9927–9936 (2017).
Laukel, M., Chistoserdova, L., Lidstrom, M. E. & Vorholt, J. A. The tungsten-containing formate dehydrogenase from Methylobacterium extorquens AM1: purification and properties. Eur. J. Biochem. 270, 325–333 (2003).
Stripp, S. T. et al. Second and outer coordination sphere effects in nitrogenase, hydrogenase, formate dehydrogenase and CO dehydrogenase. Chem. Rev. 122, 11900–11973 (2022).
Zhou, W. et al. New horizon in C1 chemistry: breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels. Chem. Soc. Rev. 48, 3193–3228 (2019).
Liew, F. et al. Gas fermentation—a flexible platform for commercial scale production of low-carbon-fuels and chemicals from waste and renewable feedstocks. Front. Microbiol. 7, 694 (2016).
Anderson, M. E., Derose, V. J., Hoffman, B. M. & Lindahl, P. A. Identification of a cyanide binding-site in CO dehydrogenase from Clostridium thermoaceticum using EPR and ENDOR spectroscopies. J. Am. Chem. Soc. 115, 12204–12205 (1993).
Shima, S. & Ataka, K. Isocyanides inhibit [Fe]-hydrogenase with very high affinity. FEBS Lett. 585, 353–356 (2011).
Kim, S. M. et al. O2-tolerant CO dehydrogenase via tunnel redesign for the removal of CO from industrial flue gas. Nat. Catal. 5, 807–817 (2022).
Kim, S. M. et al. Identifying a key spot for electron mediator-interaction to tailor CO dehydrogenase’s affinity. Nat. Commun. 15, 2732 (2024).
Dobbek, H., Svetlitchnyi, V., Gremer, L., Huber, R. & Meyer, O. Crystal structure of a carbon monoxide dehydrogenase reveals a [Ni–4Fe–5S] cluster. Science 293, 1281–1285 (2001).
Domnik, L. et al. CODH-IV: a high-efficiency CO-scavenging CO dehydrogenase with resistance to O2. Angew. Chem. Int. Ed. 56, 15466–15469 (2017).
Lee, S. H., Lee, S. M., Kang, S. G. & Lee, H. S. Enhanced H2 production by deletion of the Tfx family DNA-binding protein in the hyperthermophilic archaeon Thermococcus onnurineus NA1. Int. J. Hydrogen Energ. 46, 35189–35197 (2021).
Jang, J., Jeon, B. W. & Kim, Y. H. Bioelectrochemical conversion of CO2 to value added product formate using engineered Methylobacterium extorquens. Sci. Rep. 8, 7211 (2018).
Hartmann, T. & Leimkuhler, S. The oxygen-tolerant and NAD+-dependent formate dehydrogenase from Rhodobacter capsulatus is able to catalyze the reduction of CO2 to formate. FEBS J. 280, 6083–6096 (2013).
Choi, E. G., Yeon, Y. J., Min, K. & Kim, Y. H. Communication—CO2 reduction to formate: an electro-enzymatic approach using a formate dehydrogenase from Rhodobacter capsulatus. J. Electrochem. Soc. 165, H446–H448 (2018).
Choe, H. et al. Efficient CO2-reducing activity of NAD-dependent formate dehydrogenase from Thiobacillus sp KNK65MA for formate production from CO2 gas. PLoS One 9, e103111 (2014).
Benvenuti, M. et al. The two CO-dehydrogenases of Thermococcus sp. AM4. BBA Bioenerg. 1861, 148188 (2020).
Leo, F., Schwarz, F. M., Schuchmann, K. & Müller, V. Capture of carbon dioxide and hydrogen by engineered Escherichia coli: hydrogen-dependent CO2 reduction to formate. Appl. Microbiol. Biotechnol. 105, 5861–5872 (2021).
Kim, S. M., Kang, S. H., Jeon, B. W. & Kim, Y. H. Tunnel engineering of gas-converting enzymes for inhibitor retardation and substrate acceleration. Bioresour. Technol. 394, 130248 (2024).
Hold, C., Billerbeck, S. & Panke, S. Forward design of a complex enzyme cascade reaction. Nat. Commun. 7, 12971 (2016).
Stewart, D. W. G. et al. The role of tungsten oxide in enhancing the carbon monoxide tolerance of platinum-based hydrogen oxidation catalysts. ACS Appl. Mater. Interfaces 12, 37079–37091 (2020).
Caillat, S. Burners in the steel industry: utilization of by-product combustion gases in reheating furnaces and annealing lines. Energy Procedia 120, 20–27 (2017).
An enzyme’s new ‘tunnels’ boost its waste-grabbing prowess. Nature 609, 657 (2022).
Hwang, H. W. et al. Two-stage bioconversion of carbon monoxide to biopolymers via formate as an intermediate. Chem. Eng. J. 389, 124394 (2020).
Weissermel, K. & Arpe, H. Industrial Organic Chemistry: Important Initial and Intermediate Products (Wiley-VCH, 1976).
Liu, Q. et al. Towards a sustainable synthesis of formate salts: combined catalytic methanol dehydrogenation and bicarbonate hydrogenation. Angew. Chem. Int. Ed. 53, 7085–7088 (2014).
Calabrese, M., Russo, D., di Benedetto, A., Marotta, R. & Andreozzi, R. Formate/bicarbonate interconversion for safe hydrogen storage: a review. Renew. Sust. Energ. Rev. 173, 113102 (2023).
Eppinger, J. & Huang, K.-W. Formic acid as a hydrogen energy carrier. ACS Energy Lett. 2, 188–195 (2017).
Yishai, O., Lindner, S. N., Gonzalez de la Cruz, J., Tenenboim, H. & Bar-Even, A. The formate bio-economy. Curr. Opin. Chem. Biol. 35, 1–9 (2016).
Nakamura, M., Saeki, K. & Takahashi, Y. Hyperproduction of recombinant ferredoxins in Escherichia coli by coexpression of the ORF1-ORF2-iscS-iscU-iscA-hscB-hscA-fdx-ORF3 gene cluster. J. Biochem. 126, 10–18 (1999).
Phan, U. T., Jeon, B. W. & Kim, Y. H. Microbial engineering of Methylorubrumextorquens AM1 to enhance CO2 conversion into formate. Enzyme Microb.Technol. 168, 110264 (2023).
Miller, E., Wohlfarth, G. & Diekert, G. Studies on tetrachloroethene respiration in Dehalospirillum multivorans. Arch. Microbiol. 166, 379–387 (1996).
Moon, K., Grindstaff, J., Sobransingh, D. & Kaifer, A. E. Cueurbit[8]uril-mediated redox-controlled self-assembly of viologen-containing dendrimers. Angew.Chem. Int. Ed. 43, 5496–5499 (2004).
Tokmurzin, D. et al. High temperature flash pyrolysis characteristics of waste plastics (SRF) in a bubbling fluidized bed: effect of temperature and pelletizing. Fuel 326, 125022 (2022).
Tokmurzin, D. Three-dimensional CFD simulation of waste plastic (SRF) gasification in a bubbling fluidized bed with detailed kinetic chemical model. Energy Convers. Manag. 267, 115925 (2022).
Hamadi, N. K., Swaminathan, S. & Chen, X. D. Adsorption of paraquat dichloride from aqueous solution by activated carbon derived from used tires. J. Hazard. Mater. 112, 133–141 (2004).
Nakamura, T. et al. Adsorption removal of paraquat and diquat onto activated carbon at different adsorption temperature. Toxicol. Environ. Chem. 70, 275–280 (1999).
Lee, J. et al. Molar-scale formate production via enzymatic hydration of industrial off-gases. figshare https://doi.org/10.6084/m9.figshare.25433956.v2 (2024).
Acknowledgements
This research was supported by the C1 Gas Refinery Program and the Engineering Research Center Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT and Future Planning (2015M3D3A1A01064919 and 2020R1A5A1019631, respectively), and KEIT (1415184376).
Author information
Authors and Affiliations
Contributions
Y.H.K. and S.M.K. conceived and planned all the experiments. S.M.K. performed the bioinformatic analysis and gene cloning. S.M.K., Jinhee Lee, B.W.J., H.W.H., E.G.P. and J.K. performed the biochemical characterization, kinetic analysis and feasibility evaluation, all under the supervision of Y.H.K., S.L., H.W.R., J.N., J.-G.N. and Jinwon Lee. S.M.K. performed the structural analyses. Y.H.K. and S.M.K. wrote and reviewed the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Engineering thanks Michael Köpke and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Supplementary information
Supplementary Information
Supplementary text, Tables 1‒10, Figs. 1‒13 and References 51‒168.
Supplementary Video 1
Implementing the 10-l enzyme reactor
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, J., Kim, S.M., Jeon, B.W. et al. Molar-scale formate production via enzymatic hydration of industrial off-gases. Nat Chem Eng 1, 354–364 (2024). https://doi.org/10.1038/s44286-024-00063-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44286-024-00063-z
